Predicting DDIs in Anti-Infective Therapy: A Comprehensive Guide to PBPK Modeling Strategies
Physiologically-Based Pharmacokinetic (PBPK) modeling has emerged as a critical tool for predicting and understanding Drug-Drug Interactions (DDIs) in anti-infective therapy.
Predicting DDIs in Anti-Infective Therapy: A Comprehensive Guide to PBPK Modeling Strategies
Abstract
Physiologically-Based Pharmacokinetic (PBPK) modeling has emerged as a critical tool for predicting and understanding Drug-Drug Interactions (DDIs) in anti-infective therapy. This article provides a comprehensive overview for researchers, scientists, and drug development professionals. It first explores the foundational principles and clinical necessity of modeling DDIs involving antivirals, antifungals, and antibiotics. It then details the methodological framework for constructing and applying PBPK models, including enzyme/transporter dynamics and system parameters. The guide addresses common challenges in model development, such as parameter uncertainty and variability, and offers strategies for troubleshooting and optimization. Finally, it evaluates the validation of PBPK models against clinical DDI studies and compares their utility with traditional methods in regulatory decision-making. This synthesis aims to equip professionals with the knowledge to leverage PBPK modeling for safer and more effective combination therapies.
The Critical Need for PBPK Modeling in Anti-Infective Drug-Drug Interactions
Application Notes
Anti-infective therapy, particularly for HIV, HCV, and resistant bacterial/fungal infections, often necessitates complex multi-drug regimens. The risk for severe pharmacokinetic Drug-Drug Interactions (DDIs) is exceptionally high due to the narrow therapeutic index of many anti-infectives and their common pathways of metabolism and transport. PBPK modeling has become indispensable for de-risking development and optimizing clinical use by predicting DDI magnitudes and informing dose adjustments.
Table 1: Key DDI Mechanisms for Major Anti-Infective Classes
| Anti-Infective Class/Example | Primary DDI Mechanism | Common Interacting Drugs | Typical DDI Magnitude (AUC change) | Clinical Risk |
|---|---|---|---|---|
| HIV Protease Inhibitors (e.g., Ritonavir) | CYP3A4 Inhibition/P-gp Inhibition | Midazolam, Clarithromycin | Increase 500-1000% | Toxicity, QT prolongation |
| Non-Nucleoside Reverse Transcriptase Inhibitors (e.g., Efavirenz) | CYP3A4 Induction | Voriconazole, Rifabutin | Decrease 20-50% | Therapeutic failure |
| HCV NS5A Inhibitors (e.g., Ledipasvir) | P-gp/BCRP Substrate | Acid Reducers, Rosuvastatin | Variable (Increase/Decrease) | Altered efficacy/toxicity |
| Azole Antifungals (e.g., Voriconazole) | CYP2C19/CYP3A4 Inhibition/Substrate | Rifampin, Omeprazole | Increase 300-400% (Victim) | Toxicity or Failure |
| Fluoroquinolones (e.g., Levofloxacin) | Chelation (Cations) | Al/Mg/Fe/Ca supplements | Decrease 20-50% | Therapeutic failure |
Experimental Protocols
Protocol 1: In Vitro CYP Inhibition Assay for DDI Risk Assessment
- Objective: Determine the reversible inhibition potential (Ki) of a new anti-infective candidate on major CYP isoforms.
- Materials: Human liver microsomes (HLM), NADPH regeneration system, probe substrates (e.g., Midazolam for CYP3A4), candidate inhibitor, LC-MS/MS system.
- Procedure:
- Prepare incubation mixtures containing HLM (0.25 mg/mL), probe substrate at Km concentration, and varying concentrations of the inhibitor (e.g., 0, 0.1, 1, 10, 100 µM) in phosphate buffer.
- Pre-incubate at 37°C for 5 min. Initiate reaction by adding NADPH.
- Terminate reactions at linear time points (e.g., 5, 10, 15 min) with an organic stop solution.
- Quantify metabolite formation using LC-MS/MS.
- Analyze data using nonlinear regression to calculate IC50 and subsequently Ki.
Protocol 2: PBPK Model Development and Verification for a DDI Prediction
- Objective: Build and verify a PBPK model to predict the DDI between a novel azole (inhibitor) and a commonly co-prescribed statin (victim).
- Materials: In vitro data (logP, pKa, blood-to-plasma ratio, CYP Ki), in vivo PK data from single-ascending-dose studies, Simcyp or GastroPlus software.
- Procedure:
- System Parameters: Define a virtual population (e.g., Sim-North European Caucasian, n=10, 20-50 years).
- Compound File for Victim Drug: Populate model with physicochemical, in vitro metabolism, and plasma binding data for the statin.
- Compound File for Perpetrator: Populate with data for the azole, including its in vitro Ki and mechanism of inhibition.
- Verification: Simulate the statin's single-dose PK against clinical data. Adjust model parameters (e.g., CL, Vss) within physiological bounds to match observed data.
- DDI Prediction: Simulate the administration of the azole at steady-state with a single dose of the statin. Compare predicted vs. observed AUC and Cmax ratios.
Visualization
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in DDI Research |
|---|---|
| Pooled Human Liver Microsomes (HLM) | Contains a representative mix of human CYP enzymes for in vitro metabolism and inhibition studies. |
| Recombinant CYP Isozymes (rCYP) | Individual human CYP isoforms expressed in a standardized system for reaction phenotyping and precise Ki determination. |
| Transporter-Expressing Cell Lines (e.g., MDCK-MDR1) | Used in bidirectional assays to assess permeability and identify substrates/inhibitors of key transporters like P-gp. |
| NADPH Regeneration System | Provides essential cofactors for oxidative metabolism by CYP enzymes in microsomal incubations. |
| LC-MS/MS System with Stable Isotope Standards | Enables highly sensitive and specific quantification of drugs and their metabolites in complex biological matrices for PK analysis. |
| PBPK Software (e.g., Simcyp, GastroPlus) | Integrates in vitro, in silico, and in vivo data to build mechanistic models for human PK and DDI prediction. |
| Cryopreserved Human Hepatocytes | Provides a more physiologically complete system (including uptake transporters and basolateral efflux) for assessing intrinsic clearance and induction. |
Within the context of advancing anti-infective therapy, the management of drug-drug interactions (DDIs) is critical for therapeutic efficacy and patient safety. Physiologically Based Pharmacokinetic (PBPK) modeling has emerged as a pivotal tool for predicting complex DDIs by mechanistically simulating the ADME processes of investigational drugs and co-administered agents. These models integrate system-specific (physiological), drug-specific (physicochemical), and population-specific parameters to predict drug concentration-time profiles in plasma and tissues, enabling the in silico assessment of DDI risk prior to clinical trials.
Core Principles of ADME Simulation in PBPK
Absorption
PBPK models simulate drug absorption by representing the gastrointestinal tract as a series of physiological compartments (stomach, small intestine, colon) with distinct properties (pH, transit times, surface area, expression of transporters).
Key Model Components:
- Dissolution: Modeled using algorithms like the Johnson dissolution model, dependent on drug solubility and particle size.
- Permeability: Described via the effective intestinal permeability (Peff), often derived from Caco-2 assays or in silico predictions.
- Transporter Effects: Influx (e.g., PEPT1) and efflux (e.g., P-gp) transporters are incorporated using Michaelis-Menten kinetics.
Quantitative Parameters for Anti-Infectives: Table 1: Key Absorption Parameters for Exemplar Anti-Infective Drugs
| Drug Class | Example | Solubility (mg/mL) | Peff (×10⁻⁴ cm/s) | Transporter Involvement | Fᵃ (%) |
|---|---|---|---|---|---|
| HIV Protease Inhibitor | Ritonavir | 0.1 | 2.5 | P-gp/BCRP substrate | ~60-80 |
| Azole Antifungal | Itraconazole | 0.001 | 4.0 | CYP3A4/P-gp substrate | 55 (fasted) |
| Fluoroquinolone Antibiotic | Ciprofloxacin | 30 | 1.5 | PEPT1 influx | ~70 |
Fᵃ: Fraction absorbed
Distribution
Distribution is predicted by modeling tissue partitioning. The primary method is the permeability-limited or perfusion-limited tissue compartment model.
Key Model Components:
- Tissue-to-Plasma Partition Coefficients (Kp): Predicted using in vitro data and mechanistic models like the Poulin and Theil or Berezhkovskiy method.
- Plasma Protein Binding: Incorporates fraction unbound in plasma (fu), a critical parameter for DDI prediction.
- Specific Tissue Binding: Accounts for binding to cellular components.
Protocol 1: In Vitro Determination of Plasma Protein Binding (Ultrafiltration)
- Preparation: Spike the anti-infective drug into blank human plasma at therapeutic concentrations.
- Incubation: Incubate at 37°C for 15 minutes.
- Ultrafiltration: Load sample into a pre-rinsed centrifugal ultrafiltration device (MWCO 10 kDa).
- Centrifugation: Centrifuge at 2000 × g, 37°C, for 30 minutes.
- Analysis: Quantify drug concentration in the filtrate (unbound) and original plasma (total) using LC-MS/MS.
- Calculation: fu = Concentration(filtrate) / Concentration(plasma).
Metabolism and Excretion
PBPK models mechanistically represent metabolic pathways (via CYP enzymes, UGTs) and excretion processes (renal, biliary).
Key Model Components:
- Enzyme Kinetics: Intrinsic clearance (CLint) from human liver microsomes or hepatocytes.
- Enzyme Abundance: Tissue-specific enzyme abundance levels (pmol/mg protein).
- Transporter-Mediated Excretion: Renal (OATs, OCTs) and hepatic (OATPs, BCRP, MRP2) transporters using kinetic parameters (Vmax, Km).
Quantitative Data for DDI Prediction: Table 2: Key Disposition Parameters for Anti-Infective DDI Modeling
| Process | Parameter | Symbol | Value (Example - Ritonavir) | Source |
|---|---|---|---|---|
| Metabolism | CYP3A4 CLint | CLint,u | 0.5 µL/min/pmol | HLM assay |
| CYP3A4 Inhibition | Kᵢ | 0.02 µM (potent) | Recombinant enzyme | |
| Biliary Excretion | P-gp Vmax (Liver) | Vmax | 500 pmol/min/mg protein | Transfected cell assay |
| P-gp Km | Km | 200 µM | Transfected cell assay | |
| Renal Excretion | Fraction Unchanged in Urine | fe | 0.11 | Clinical data |
| Glomerular Filtration Rate | GFR | 120 mL/min | System parameter |
Protocol 2: Determining Time-Dependent CYP3A4 Inhibition (TDI) Parameters for PBPK
- Incubation: Pre-incubate human liver microsomes (0.5 mg/mL) with the anti-infective drug (multiple concentrations) and NADPH for 0-30 min.
- Dilution: Dilute aliquots 20-fold into a secondary incubation containing a specific CYP3A4 probe substrate (e.g., midazolam) and NADPH.
- Reaction Termination: Stop secondary reaction with acetonitrile at timed intervals.
- Analysis: Quantify metabolite formation (1'-OH-midazolam) via LC-MS/MS.
- Data Fitting: Fit depletion data to determine kinact (maximum inactivation rate) and KI (inactivator concentration for half-maximal inactivation).
Integration for DDI Prediction
For a DDI simulation between a perpetrator (e.g., ritonavir) and victim drug (e.g., a CYP3A4 substrate), the model simultaneously simulates the perpetrator's concentration-time profile in the liver and gut, which then modulates the enzyme/transporter activity (via Ki or kinact/KI) for the victim drug, altering its ADME profile.
PBPK Model Development and DDI Prediction Workflow
Interplay of ADME Processes in a PBPK Framework
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for PBPK-Relevant In Vitro ADME Assays
| Item | Function in PBPK Context | Example Product/Catalog |
|---|---|---|
| Cryopreserved Human Hepatocytes | Source for determining intrinsic clearance (CLint), metabolite identification, and induction studies. Critical for scaling hepatic metabolism. | BioIVT Human Hepatocytes, Lot-specific. |
| Human Liver Microsomes (HLM) | Used for determining cytochrome P450 enzyme kinetic (Vmax, Km) and inhibition (Ki) parameters. | Corning Gentest UltraPool HLM 150-donor. |
| Recombinant CYP Enzymes | For reaction phenotyping to identify which specific CYP isoform metabolizes a drug. | BD Supersomes (CYP3A4, 2D6, etc.). |
| Caco-2 Cell Line | Standard in vitro model for predicting human intestinal permeability (Peff) and studying transporter effects (P-gp). | ATCC HTB-37. |
| Transfected Cell Systems (OATP1B1, P-gp, etc.) | Used to quantify transporter-specific uptake/efflux kinetics (Vmax, Km) for hepatic and renal clearance models. | Solvo MDCKII-MDR1, ThermoFisher Flp-In-293-OATP1B1. |
| LC-MS/MS System | Quantitative bioanalysis for measuring drug concentrations in in vitro assays and in vivo samples (plasma, tissues). | SCIEX Triple Quad 6500+, Waters Xevo TQ-S. |
| PBPK Software Platform | Integrated environment for building, simulating, and validating PBPK models (e.g., Simcyp Simulator, GastroPlus, PK-Sim). | Certara Simcyp Simulator v22. |
Application Notes
Drug-drug interactions (DDIs) are a critical determinant of therapeutic success and toxicity in anti-infective therapy. Mechanistic understanding of DDIs via Cytochrome P450 (CYP450) enzymes, transporters, and plasma protein binding is essential for dose optimization. Within Physiologically Based Pharmacokinetic (PBPK) modeling, these mechanisms are quantified to predict alterations in drug exposure, informing clinical decision-making and drug labeling.
1.1. CYP450 Enzyme-Mediated Interactions Anti-infectives can act as perpetrators (inhibitors/inducers) or victims (substrates) of CYP enzymes. Macrolides (e.g., clarithromycin) are potent mechanism-based inhibitors of CYP3A4, drastically increasing exposure to co-administered drugs like rifabutin, leading to uveitis. Rifampin, a broad-spectrum inducer of CYP3A4, CYP2C9, and others, decreases exposure to victim drugs like voriconazole and protease inhibitors, risking therapeutic failure. PBPK models integrate in vitro parameters (e.g., Ki, kinact) to simulate these time- and concentration-dependent effects.
1.2. Transporter-Mediated Interactions Hepatic (OATP1B1/1B3) and renal (OATs, OCTs, MATEs) transporters govern the distribution and clearance of many anti-infectives. Coadministration of the OATP1B1 inhibitor cyclosporine with the substrate rifampin increases rifampin AUC by ~250%, elevating hepatotoxicity risk. The combination of tenofovir (substrate of renal OATs and MRP4) with cobicistat (inhibitor of MATE1) reduces tenofovir secretion, increasing plasma levels and potential for nephrotoxicity. PBPK models require transporter expression data, inhibition constants (IC50), and fractional transport contributions (ft) for accurate prediction.
1.3. Protein Binding Displacement Displacement from plasma proteins (e.g., albumin, alpha-1-acid glycoprotein) can cause transient increases in free, pharmacologically active drug concentration. While often clinically insignificant due to compensatory clearance mechanisms, it can be critical for drugs with high extraction ratio, narrow therapeutic index, and high protein binding (>95%). Ceftriaxone displaces bilirubin, risking kernicterus in neonates. For highly bound drugs like itraconazole (>99% bound), displacement interactions require careful PBPK characterization of free drug concentration.
1.4. Quantitative DDI Data Summary for Key Anti-Infectives
Table 1: Key Perpetrator Anti-Infectives and Their DDI Magnitude
| Perpetrator Drug | Mechanism | Victim Drug | Change in Victim AUC | Clinical Risk |
|---|---|---|---|---|
| Clarithromycin | CYP3A4 Inhibition | Rifabutin | Increase ~400% | Uveitis, Neutropenia |
| Rifampin | CYP3A4 Induction | Voriconazole | Decrease ~90% | Therapeutic Failure |
| Ritonavir/Cobicistat | CYP3A4 + Transporter Inhibition | Atorvastatin | Increase ~500% | Myopathy |
| Cyclosporine | OATP1B1 Inhibition | Rifampin | Increase ~250% | Hepatotoxicity |
| Probenecid | OAT1/3 Inhibition | Cephalexin | Increase ~300% | CNS Toxicity |
Table 2: Key Victim Anti-Infectives and Their Vulnerability
| Victim Drug | Primary Clearance Pathway | Key Perpetrator | Change in Anti-Infective AUC | Recommendation |
|---|---|---|---|---|
| Isavuconazole | CYP3A4 Metabolism | Rifampin (Inducer) | Decrease ~90% | Contraindicated |
| Telithromycin | CYP3A4 Metabolism | Ketoconazole (Inhibitor) | Increase ~250% | Dose Adjustment |
| Tenofovir DF | Renal (OATs, MATEs) | Cobicistat (MATE1 Inhib.) | Increase ~30-40% | Monitor Renal Function |
| Dalbavancin | Non-enzymatic, Protein Binding | Warfarin (Displacement) | Minimal Change in Free | Monitor INR |
Experimental Protocols
2.1. Protocol for Determining CYP450 Inhibition Kinetics (Time-Dependent Inhibition) Objective: To characterize the kinetics of mechanism-based inhibition (MBI) of a CYP enzyme (e.g., CYP3A4) by a test anti-infective (e.g., clarithromycin).
- Materials: Human liver microsomes (HLM, 0.5 mg/mL), NADPH regeneration system, CYP3A4-specific probe substrate (midazolam, 2.5 µM), test inhibitor (clarithromycin, 0-100 µM), potassium phosphate buffer (100 mM, pH 7.4), LC-MS/MS system.
- Primary Incubation: Pre-incubate HLM with test inhibitor (multiple concentrations) and NADPH in buffer at 37°C. Include control without NADPH.
- Sampling: At pre-determined time points (0, 5, 10, 20, 30 min), remove aliquots from the primary incubation mix.
- Secondary Incubation: Dilute each aliquot 20-fold into a secondary incubation containing the probe substrate (midazolam) and NADPH to measure remaining CYP3A4 activity.
- Reaction Termination: Stop secondary incubation after 5 min with cold acetonitrile containing internal standard.
- Analysis: Quantify metabolite formation (1'-hydroxymidazolam) via LC-MS/MS.
- Data Analysis: Plot natural log of remaining activity vs. pre-incubation time for each inhibitor concentration. The slope = kobs. Plot kobs vs. inhibitor concentration [I] to determine kinact (maximal inactivation rate) and KI (inhibitor concentration for half-maximal inactivation). Use equation: kobs = (kinact * [I]) / (KI + [I]).
2.2. Protocol for OATP1B1 Uptake Assay in Transfected Cells Objective: To assess if a new anti-infective is a substrate or inhibitor of the OATP1B1 transporter.
- Materials: HEK293 cells stably expressing OATP1B1 and mock-transfected controls, reference substrate ([³H]-estradiol-17β-D-glucuronide, E17βG), test compound, uptake buffer (Hanks' Balanced Salt Solution, HBSS, pH 7.4), liquid scintillation counter or LC-MS/MS.
- Cell Preparation: Seed cells in 24-well plates 48h prior. Wash monolayers twice with warm HBSS.
- Uptake Phase: Incubate cells with E17βG (tracer concentration, e.g., 1 µM) ± test inhibitor (various concentrations) or with test compound as potential substrate. Perform incubations at 37°C for a predetermined time (e.g., 2-5 min).
- Termination: Rapidly wash wells 3x with ice-cold HBSS. Lyse cells with 0.1% Triton X-100 or NaOH.
- Quantification: Measure radioactivity via scintillation counting or analyze lysate by LC-MS/MS for test compound.
- Data Analysis: For inhibition: Calculate IC50 by fitting inhibition data to a logistic model. For substrate identification: Compare uptake in OATP1B1 vs. mock cells (≥2-fold difference indicates substrate activity).
2.3. Protocol for Determining Plasma Protein Binding via Equilibrium Dialysis Objective: To measure the fraction unbound (fu) of a highly protein-bound anti-infective in human plasma.
- Materials: Human plasma (heparinized), equilibrium dialysis device (e.g., RED plate), dialysis membrane (molecular weight cutoff ~12-14 kDa), phosphate buffer (pH 7.4), test anti-infective, humidified incubator (37°C, 5% CO2).
- Setup: Spike test drug into plasma to a therapeutic concentration (e.g., 10 µg/mL). Load plasma into the donor chamber and buffer into the receiver chamber.
- Dialysis: Seal plate and incubate with gentle agitation for 4-6 hours to reach equilibrium.
- Sampling: Post-dialysis, sample from both plasma and buffer chambers.
- Analysis: Quantify drug concentration in both matrices using a validated bioanalytical method (LC-MS/MS).
- Calculation: Calculate fraction unbound: fu = [Drug]buffer / [Drug]plasma. Ensure mass balance is within 80-120%.
Visualizations
Title: PBPK Modeling Integrates Three Key DDI Mechanisms
Title: Experimental Protocol for Time-Dependent CYP Inhibition
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for DDI Mechanistic Studies
| Reagent/Material | Function/Application | Example Vendor/Product |
|---|---|---|
| Pooled Human Liver Microsomes (HLM) | In vitro system containing CYP450 enzymes for metabolism and inhibition studies. | Corning Gentest, Xenotech |
| Recombinant CYP Enzymes (rCYP) | Individual, expressed CYP isoforms (e.g., rCYP3A4) for reaction phenotyping. | BD Biosciences, Cypex |
| Transporter-Transfected Cell Lines | Cells overexpressing a single human transporter (e.g., OATP1B1-HEK) for uptake studies. | Solvo Biotechnology, GenoMembrane |
| Caco-2 Cell Monolayers | Model for intestinal permeability and P-gp/BCRP efflux transporter studies. | ATCC, ECACC |
| Equilibrium Dialysis Devices | Gold-standard method for determining plasma protein binding (fu). | Thermo Fisher (RED), HTDialysis |
| LC-MS/MS System | High-sensitivity quantification of drugs and metabolites in complex biological matrices. | Sciex, Agilent, Waters |
| NADPH Regeneration System | Supplies essential cofactor for CYP450 and some reductase enzyme reactions. | Promega, Sigma-Aldrich |
| Stable Isotope-Labeled Internal Standards | Ensures accuracy and precision in bioanalytical quantification by compensating for matrix effects. | Cerilliant, Toronto Research Chemicals |
Application Notes on PBPK Modeling for Anti-Infective DDIs
Physiologically-based pharmacokinetic (PBPK) modeling is a critical tool for predicting and managing drug-drug interactions (DDIs) in complex anti-infective regimens. This is especially vital in the high-risk scenarios of HIV, Hepatitis C, systemic fungal infections, and polypharmacy, where co-infections and comorbidities are common. PBPK models integrate system-specific (physiological) and drug-specific (physicochemical, pharmacokinetic) parameters to mechanistically simulate drug exposure, enabling the prediction of DDI magnitude prior to clinical studies. This supports dose optimization, risk assessment for therapeutic failure or toxicity, and the design of informed clinical DDI trials.
HIV (HAART) - Key DDI Mechanisms
HAART regimens, particularly those containing pharmacokinetic enhancers (e.g., ritonavir, cobicistat), are prone to causing DDIs via potent inhibition of cytochrome P450 3A4 (CYP3A4) and P-glycoprotein (P-gp). Concurrently, non-nucleoside reverse transcriptase inhibitors (e.g., efavirenz) are inducers of CYP enzymes. These opposing effects create a complex DDI landscape when managing co-infections.
Hepatitis C - Direct-Acting Antivirals (DAAs)
While modern DAAs are generally safer than older interferons, they are significant substrates and moderates inhibitors of drug transporters (P-gp, BCRP, OATP1B1/1B3) and CYP enzymes. Co-administration with HAART or azole antifungals requires careful evaluation due to overlapping metabolic pathways.
Systemic Fungal Infections - Azole Antifungals
Triazole antifungals (e.g., voriconazole, itraconazole, posaconazole) are potent inhibitors of CYP3A4 and are themselves substrates of CYP2C19 and CYP3A4. Their long half-lives and nonlinear pharmacokinetics complicate DDI management. They represent a high risk for increasing exposure to co-administered drugs.
Polypharmacy in Co-infected Patients
Patients with HIV/HCV co-infection or those with opportunistic fungal infections often receive 5+ medications. This polypharmacy exponentially increases DDI risks due to cumulative effects on shared metabolic and transporter pathways, leading to altered drug exposure, increased toxicity, or loss of efficacy.
Table 1: Representative High-Risk DDI Magnitudes in Anti-Infective Therapy
| Victim Drug (Object) | Perpetrator Drug (Precipitant) | Interaction Mechanism | Change in AUC (Mean Ratio) | Clinical Risk |
|---|---|---|---|---|
| Maraviroc (CCR5 antagonist) | Ritonavir (PI booster) | CYP3A4/P-gp inhibition | Increase: ~ 9.5-fold | Potential for toxicity (hepatotoxicity) |
| Tenofovir alafenamide | Cobicistat | P-gp/BCRP inhibition | Increase: ~ 1.3-1.4 fold | Monitoring for renal effects |
| Sofosbuvir (HCV NS5B inhibitor) | Rifampin (for TB) | P-gp induction | Decrease: ~ 0.5-fold | Risk of therapeutic failure |
| Voriconazole (Azole) | Efavirenz (NNRTI) | CYP2C19/CYP3A4 induction | Decrease: ~ 0.4-fold | Loss of antifungal efficacy |
| Atazanavir (PI) | Omeprazole (PPI) | Increased gastric pH | Decrease: ~ 0.3-fold | Reduced ARV absorption & efficacy |
| Ledipasvir (HCV NS5A inhibitor) | Rosuvastatin | OATP1B1/BCRP inhibition | Increase: ~ 5.6-fold (rosuvastatin AUC) | Increased statin toxicity risk |
Detailed Experimental Protocols
Protocol 1:In VitroCYP Inhibition Assay for DDI Risk Assessment
Purpose: To determine the reversible inhibition potential (IC50/Ki) of a new anti-infective drug against major CYP isoforms (3A4, 2D6, 2C9, 2C19, 1A2). Materials:
- Recombinant human CYP enzymes or human liver microsomes (HLM).
- CYP-specific probe substrates (see Reagent Solutions table).
- Co-factor: NADPH regeneration system.
- Stop reagent: Acetonitrile with internal standard.
- LC-MS/MS system for analysis. Procedure:
- Prepare incubation mixtures (final volume 100 µL) containing phosphate buffer (pH 7.4), HLM (0.1 mg/mL), and the test anti-infective drug at 8 concentrations (e.g., 0.1-100 µM).
- Pre-incubate at 37°C for 5 min. Initiate reaction by adding NADPH and probe substrate at Km concentration.
- Incubate for a linear time period (e.g., 10 min). Terminate reaction with ice-cold acetonitrile.
- Centrifuge, analyze supernatant via LC-MS/MS to quantify metabolite formation from the probe.
- Calculate % activity remaining vs. control (no inhibitor). Plot dose-response curve to determine IC50. Calculate Ki using the Cheng-Prusoff equation if needed.
Protocol 2: PBPK Model Development and Verification for a DDI Study
Purpose: To build and verify a PBPK model predicting the DDI between a new azole antifungal and a boosted HIV protease inhibitor. Materials:
- PBPK software (e.g., Simcyp Simulator, GastroPlus, PK-Sim).
- In vitro ADME data for both drugs: logP, pKa, blood-to-plasma ratio, fu, Km/Vmax for relevant CYPs, CLint.
- Clinical PK data (single/multiple dose) for each drug alone.
- Demographic data for virtual population (e.g., Sim-Healthy Volunteers). Procedure:
- Model Drug (Perpetrator): Develop a full-PBPK model for the azole antifungal. Input physicochemical and in vitro data. Optimize model parameters (e.g., CLint, Vss) by fitting to clinical PK data.
- Model Drug (Victim): Develop a minimal-PBPK (compartmental) model for the protease inhibitor. Incorporate known metabolic pathways (CYP3A4). Verify model against clinical PK data.
- Implement DDI Mechanism: In the software, define the azole as a reversible inhibitor of CYP3A4 using its in vitro Ki value. For mechanism-based inhibition, input kinact and KI.
- Design Virtual Trial: Simulate a clinical DDI study: N=100 (10 trials x 10 subjects), matching the age, sex, and genotype of a reference study. Administer victim drug alone, then co-administer with perpetrator at steady-state.
- Verify Model: Compare simulated AUC and Cmax ratios (with/without inhibitor) against observed data from a published clinical DDI study. Accept if predicted/observed ratios fall within 1.5-fold.
- Probe Extrapolation: Use the verified model to simulate DDI risk in special populations (e.g., hepatic impairment) or with different dosing regimens.
Diagrams
Diagram Title: Key Pharmacokinetic DDI Pathways for Anti-Infectives
Diagram Title: PBPK Model Development and DDI Prediction Steps
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for In Vitro DDI Studies
| Item | Function/Application | Example Product/Kit |
|---|---|---|
| Human Liver Microsomes (HLM) | Pooled subcellular fraction containing CYP enzymes; used for reaction phenotyping and inhibition assays. | Corning Gentest, XenoTech HLM. |
| Recombinant Human CYP Enzymes | Individual CYP isoforms (baculovirus-expressed); used for specific reaction phenotyping. | Corning Supersomes. |
| CYP-Specific Probe Substrates | Selective substrates metabolized to a quantifiable product by a single CYP isoform. | Midazolam (CYP3A4), Bupropion (CYP2B6), Diclofenac (CYP2C9). |
| NADPH Regeneration System | Provides reducing equivalents (NADPH) essential for CYP enzymatic activity. | Sigma-Aldrich NADPH Regenerating System Solution A & B. |
| Caco-2 Cells | Human colon adenocarcinoma cell line forming polarized monolayers; gold standard for in vitro permeability and P-gp transport studies. | ATCC HTB-37. |
| Transfected Cell Systems | Cells overexpressing a single transporter (e.g., MDCKII-MDR1, HEK-OATP1B1) for specific uptake/efflux assays. | Solvo Biotechnology Transporter Assay Kits. |
| LC-MS/MS System | High-sensitivity analytical platform for quantifying drugs and metabolites in biological matrices from in vitro and in vivo studies. | SCIEX Triple Quad, Agilent 6470. |
| PBPK Simulation Software | Platform for mechanistic modeling of ADME and DDI predictions. | Certara Simcyp Simulator, Simulations Plus GastroPlus. |
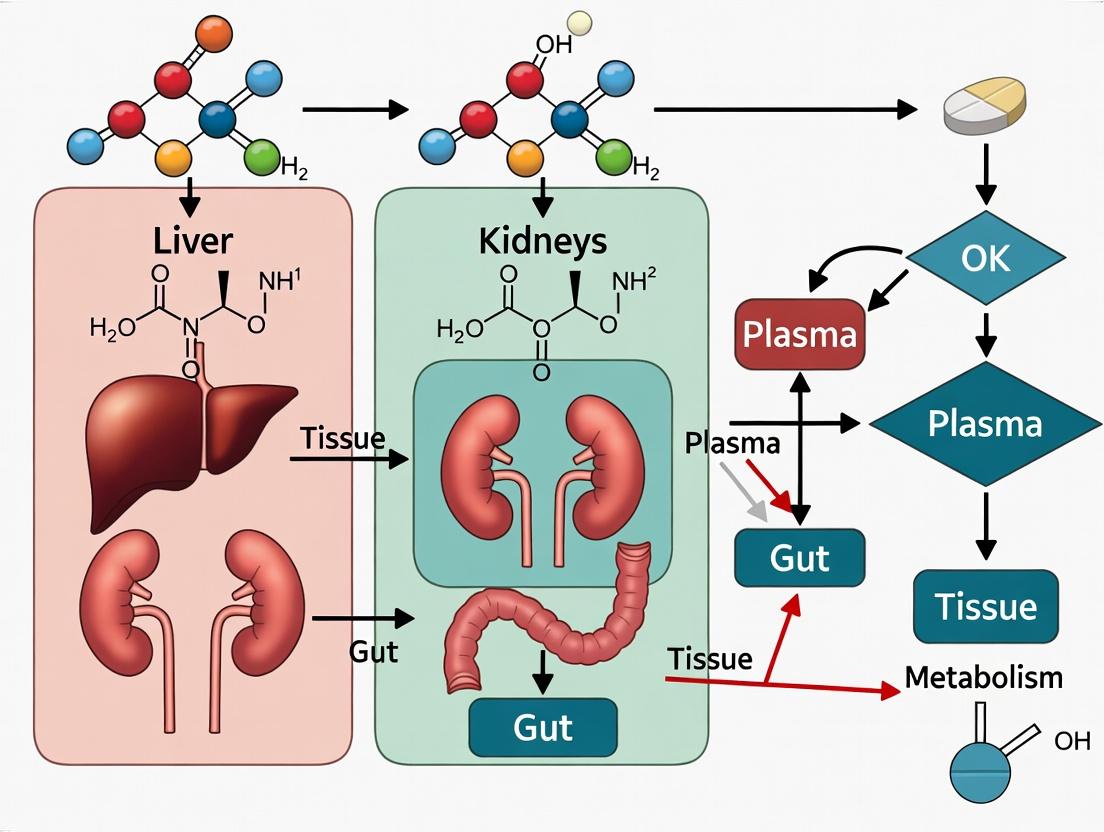
Within the broader thesis on advancing Physiologically-Based Pharmacokinetic (PBPK) modeling for drug-drug interaction (DDI) prediction in anti-infective therapy, this Application Note examines the regulatory paradigm shift. Anti-infectives (e.g., antiretrovirals, antifungals, antimycobacterials) are frequently perpetrators and victims of DDIs due to potent inhibition/induction of cytochrome P450 enzymes and transporters. Regulatory agencies now formally encourage PBPK to optimize clinical DDI assessment strategies.
The following table synthesizes key quantitative and qualitative recommendations from the latest FDA (2020) and EMA (2021/2022) guidance documents.
Table 1: Comparison of FDA and EMA Guideline Positions on PBPK for DDI Assessment
| Aspect | U.S. FDA Guidance for Industry: "Clinical Drug Interaction Studies" (2020) | EMA Guideline on the Qualification and Reporting of PBPK Modelling (2021) & DDI Guideline (2012, revised 2023) |
|---|---|---|
| Primary Stance | Explicitly "encourages" the use of PBPK modeling. | "Supports" and "recommends" PBPK approaches as part of drug development. |
| Key Application: Victim DDI | To support waiver for a clinical study when model predicts AUC ratio ≤ threshold (e.g., ≤ 1.25 for strong inhibitors). | To justify a waiver for in vivo DDI studies, especially when risk is predicted to be low. |
| Key Application: Perpetrator DDI | To optimize design of clinical DDI studies (e.g., dosing regimen, subject selection). To support alternative dosing strategies. | To replace a clinical study for weak inhibitors/inducers if justified by a validated model. |
| Model Validation | Requires verification against clinical PK/DDI data. "Prior experience" with the platform can support credibility. | Stresses "qualification" of the platform and model. Internal (development data) and external (literature) validation are critical. |
| Substrate Specificity | Discusses use for enzyme (CYP) and transporter (P-gp, BCRP, OATP1B1/1B3) mediated DDIs. | Similarly covers enzymes and transporters. Strong emphasis on transporter DDI assessment. |
| Reporting Standards | Detailed documentation of model inputs, assumptions, and verification steps is required in regulatory submissions. | Requires comprehensive reporting per the EMA PBPK guideline template, including sensitivity analyses. |
Application Note: PBPK to Inform DDI Strategy for a Novel Antifungal
Scenario: Development of a novel CYP3A4 substrate antifungal drug (Drug S) with potential for concomitant use with a strong CYP3A4 inhibitor (Drug I).
Aim: To use PBPK modeling to assess the need for, or design of, a clinical DDI study.
Workflow Diagram:
Diagram Title: PBPK Workflow for DDI Assessment & Regulatory Strategy
Experimental Protocols for In Vitro Data Generation for PBPK
High-quality in vitro data are critical PBPK inputs. Below are detailed protocols for key experiments.
Protocol 4.1: Determination of Metabolic Stability & Intrinsic Clearance (CLint) in Human Liver Microsomes (HLM)
Objective: To quantify the in vitro degradation half-life (t1/2) and calculate CLint for primary metabolic pathways. Materials: See "The Scientist's Toolkit" (Section 6). Procedure:
- Prepare incubation mixture (final 0.1 mg/mL HLM, 1 μM test drug in 100 mM phosphate buffer, pH 7.4).
- Pre-incubate mixture at 37°C for 5 min.
- Initiate reaction by adding NADPH regenerating system (final 1 mM NADP+, 5 mM G6P, 1 U/mL G6PDH).
- At predefined times (0, 5, 10, 20, 30, 45 min), aliquot 50 μL into 150 μL of stop solution (acetonitrile with internal standard).
- Centrifuge (4000xg, 15 min, 4°C) and analyze supernatant via LC-MS/MS.
- Plot natural log of parent drug remaining vs. time. Calculate k (slope), in vitro t1/2 = 0.693/k.
- Calculate CLint (μL/min/mg protein) = (0.693 / t1/2) * (Incubation Volume / Protein Amount).
Protocol 4.2: Determination of Inhibition Constant (Ki) for a CYP Enzyme
Objective: To determine the Ki value of a perpetrator drug (I) against a specific CYP isoform (e.g., CYP3A4). Procedure:
- Select a probe substrate (S) for the target CYP (e.g., midazolam for CYP3A4).
- Perform incubations with HLM at six concentrations of I (spanning expected range around IC50) and six concentrations of S (around its Km).
- Follow Protocol 4.1 steps for incubation and analysis, measuring metabolite formation rate (v).
- Fit data using nonlinear regression to appropriate inhibition model (competitive, mixed, non-competitive) using software (e.g., Phoenix WinNonlin). The model yielding the lowest AIC value is typically selected.
- Report Ki value and the preferred inhibition mechanism.
Pathway Diagram: DDI Mechanism for a Common Anti-infective Scenario
Diagram Title: CYP3A4 Inhibition DDI in Gut and Liver
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for In Vitro DDI-PBPK Assays
| Item | Function in PBPK Context | Example Product / Source |
|---|---|---|
| Pooled Human Liver Microsomes (HLM) | Contains the full complement of human CYP enzymes for measuring metabolic CLint and inhibition. | XenoTech HMMCPL, Corning Gentest Pooled HLM |
| Recombinant CYP Enzymes (rCYP) | Isoform-specific reaction phenotyping to attribute fraction metabolized (fm) by each pathway. | BD Supersomes (CYP3A4, 2D6, etc.) |
| Transporter-Expressing Cell Lines | To assess substrate/inhibition potential for key transporters (P-gp, BCRP, OATPs). | MDCKII-MDR1, HEK293-OATP1B1 |
| NADPH Regenerating System | Provides essential cofactor for oxidative metabolism in microsomal incubations. | Corning Gentest NADP Regenerating System |
| LC-MS/MS System | Gold-standard for quantitative analysis of drugs and metabolites in complex in vitro matrices. | SCIEX Triple Quad systems, Agilent HPLC/QQQ |
| PBPK Software Platform | Integrates in vitro data and system parameters to build, simulate, and validate models. | Simcyp Simulator, GastroPlus, PK-Sim |
| Specific Chemical Inhibitors | Positive controls for reaction phenotyping and inhibition studies (e.g., Ketoconazole for CYP3A4). | Available from multiple chemical suppliers (e.g., Sigma-Aldrich) |
Building and Applying PBPK Models for DDI Prediction: A Step-by-Step Framework
Within the broader thesis on PBPK (Physiologically Based Pharmacokinetic) modeling for drug-drug interactions (DDIs) in anti-infective therapy research, establishing a robust and standardized model development workflow is paramount. Anti-infectives, including antivirals, antibacterials, and antifungals, are frequently co-administered with other medications in complex patient populations, elevating DDI risk. This application note details a comprehensive, iterative workflow from initial compound data entry to virtual population simulation, ensuring predictive, reliable DDI assessments for anti-infective drugs.
Application Notes: Core Workflow Stages
Stage 1: Compound Data Entry and Parameterization
This initial stage involves the systematic collection and entry of compound-specific physicochemical and in vitro data. The accuracy of this foundational dataset directly influences model predictability.
Key Data Requirements:
- Physicochemical Properties: Molecular weight, logP, pKa, solubility, blood-to-plasma ratio.
- Binding Data: Fraction unbound in plasma (fu) and in microsomes (fu,mic).
- In Vitro Metabolism & Transport: Intrinsic clearance (CLint), Michaelis-Menten constants (Km, Vmax) for relevant Cytochrome P450 (CYP) enzymes and drug transporters (e.g., P-gp, OATP1B1/1B3). Inhibitory constants (Ki, IC50) for DDIs.
- Permeability: Caco-2 or Papp values.
Table 1: Example Quantitative Data Entry for a Hypothetical Antiviral (Drug A) and a CYP3A4 Inhibitor (Drug B)
| Parameter | Symbol | Drug A (Victim) | Drug B (Perpetrator) | Source Experiment |
|---|---|---|---|---|
| Molecular Weight (g/mol) | MW | 450.5 | 350.2 | QC-MS |
| Log Partition Coefficient | LogP | 2.1 | 3.8 | Shake-flask |
| Acid Dissociation Constant | pKa (acid) | 4.5 | N/A | Potentiometry |
| Fraction Unbound (Plasma) | fu | 0.15 | 0.02 | Equilibrium Dialysis |
| CYP3A4 CLint (µL/min/pmol) | CLint, 3A4 | 2.5 | N/A | Human Liver Microsomes |
| CYP3A4 Inhibition Constant (µM) | Ki | N/A | 0.15 | Recombinant CYP3A4 |
Protocol 2.1: Determination of Fraction Unbound in Plasma (fu) via Equilibrium Dialysis Objective: To measure the unbound fraction of a drug in human plasma. Materials: See Scientist's Toolkit. Procedure:
- Prepare a solution of the test compound in phosphate-buffered saline (PBS, pH 7.4) and spike into human plasma to achieve a therapeutically relevant concentration (e.g., 1 µM).
- Load the plasma sample into one chamber of a pre-hydrated equilibrium dialysis device. Load PBS into the opposing chamber.
- Seal the device and incubate at 37°C with gentle agitation for 4-6 hours to reach equilibrium.
- Post-incubation, collect aliquots from both the plasma and buffer chambers.
- Quench samples with an equal volume of acetonitrile containing an internal standard. Vortex and centrifuge (4000xg, 15 min) to precipitate proteins.
- Analyze the supernatant using a validated LC-MS/MS method to determine compound concentrations in the plasma ([C]plasma) and buffer ([C]buffer) chambers.
- Calculate fu: fu = [C]buffer / [C]plasma. Correct for any volume shift.
Stage 2:In VitrotoIn VivoExtrapolation (IVIVE)
This stage translates the in vitro parameters (e.g., CLint) into in vivo physiological scales (e.g., hepatic clearance, CLh).
Core Calculations:
- Hepatic Clearance: Using the "well-stirred" liver model: CLh = (Qh * fu * CLint) / (Qh + fu * CLint), where Qh is hepatic blood flow (~20 mL/min/kg).
- Renal & Biliary Clearance: Estimated from in vitro transporter data or in vivo recovery studies.
Stage 3: Base Model Building and Verification
A minimal PBPK model (often a whole-body or simplified compartmental model) is built using specialized software (e.g., GastroPlus, Simcyp Simulator, PK-Sim). The model incorporates compound parameters and system (physiological) parameters. The model is verified by comparing its simulations to observed single-agent pharmacokinetic (PK) data from Phase I clinical trials.
Table 2: Model Verification Metrics for Drug A Base Model
| PK Parameter | Observed Geometric Mean | Simulated Geometric Mean | Prediction Error (%) | Acceptance Criteria |
|---|---|---|---|---|
| Cmax (ng/mL) | 1250 | 1187 | -5.0% | ±20% |
| AUC0-∞ (ng·h/mL) | 8500 | 8925 | +5.0% | ±20% |
| t1/2 (h) | 12.0 | 11.3 | -5.8% | ±30% |
Stage 4: DDI Mechanism Integration and Validation
Mechanisms of interaction (e.g., competitive CYP inhibition, induction, transporter inhibition) are integrated. For the anti-infective DDI thesis, this is the critical step. The DDI model is validated against clinical DDI studies.
Protocol 2.4: Modeling Competitive CYP Inhibition DDI Objective: To simulate the effect of a perpetrator (Drug B) on the exposure of a victim (Drug A) metabolized by CYP3A4. Methodology:
- In the software, define Drug B's inhibitory parameters (Ki or IC50, mechanism) against CYP3A4.
- Define the enzyme kinetics (Km, Vmax/ISEF) for Drug A's clearance via CYP3A4 in the system file.
- For the DDI simulation, implement an interaction model where the perpetrator increases the apparent Km of the victim according to the equation: Km,app = Km * (1 + [I]/Ki), where [I] is the relevant inhibitor concentration at the enzyme site (e.g., hepatic inlet, maximum systemic).
- Run a simulation of the victim administered alone and then co-administered with the perpetrator.
- Calculate the predicted DDI ratio (AUCwith inhibitor / AUCalone) and compare to the observed clinical ratio.
Stage 5: Virtual Population Simulation
The final validated model is used to simulate PK and DDI outcomes in virtual populations, reflecting real-world variability. This is essential for anti-infective therapy, where patient factors (age, organ function, genetics, co-medications) vary widely.
Key Simulation Steps:
- Define Population: Select a virtual population (e.g., Simcyp's "Virtual European Population," n=100, 10 trials).
- Set Trial Design: Mirror the clinical scenario of interest (e.g., "Drug A 200mg BID for 7 days, with Drug B 400mg QD added on Day 4").
- Execute Simulation: Run the virtual clinical trial.
- Analyze Output: Generate predicted concentration-time profiles, AUC and Cmax distributions, and DDI risk stratification (e.g., percentage of virtual subjects with AUC increase >2-fold).
Visualizations
Diagram 1: PBPK Model Development & DDI Assessment Workflow
Diagram 2: Key CYP Inhibition DDI Mechanism in Liver
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for PBPK Model Development
| Item | Function in Workflow | Example Product/Source |
|---|---|---|
| Human Liver Microsomes (HLM) | Pooled in vitro system to determine metabolic stability (CLint) and enzyme kinetic parameters (Km, Vmax). | Corning Gentest, XenoTech |
| Recombinant CYP Enzymes | Isozyme-specific reaction phenotyping to identify primary metabolic pathways. | Baculovirus-insect cell expressed CYP (Supersomes). |
| Transfected Cell Systems | Assessment of drug transporter (e.g., P-gp, OATP) kinetics (uptake/Vmax, Km) and inhibition. | MDCK/MDCKII, HEK293 cells overexpressing human transporters. |
| Human Plasma (Pooled) | Determination of plasma protein binding (fu) via equilibrium dialysis or ultrafiltration. | Commercial bio-banks (e.g., BioIVT). |
| Equilibrium Dialysis Device | Gold-standard method for separating protein-bound and unbound drug fractions. | HTD96b dialysis blocks (HTDialysis), RED plates. |
| PBPK/PD Modeling Software | Platform to integrate compound and system data, perform IVIVE, build models, and run simulations. | Simcyp Simulator, GastroPlus, PK-Sim. |
| LC-MS/MS System | Quantitative bioanalysis for measuring drug concentrations in in vitro assays and in vivo samples (for verification). | Triple quadrupole mass spectrometers (e.g., SCIEX, Agilent, Waters). |
Application Notes: PBPK Modeling of Anti-Infective DDIs
Within the framework of a thesis on advancing PBPK modeling for anti-infective therapy, the accurate in silico prediction of drug-drug interactions (DDIs) is paramount. Anti-infectives are often both victims of metabolism/transport and perpetrators of enzyme/transporter modulation, leading to complex DDI networks. This document outlines the core principles and protocols for characterizing perpetrator (inhibitor/inducer) and victim drug kinetics, which form the foundation for robust whole-body PBPK model construction and DDI simulation.
1. Quantitative Parameters for DDI PBPK Modeling The following parameters must be obtained in vitro and scaled to the in vivo context for both perpetrator and victim drugs.
Table 1: Essential Victim Drug Parameters for Enzyme-Mediated Clearance
| Parameter | Symbol | Typical Units | Description & Relevance |
|---|---|---|---|
| Fraction metabolized by CYP enzyme | fm,CYP | Unitless | Fraction of total clearance via a specific CYP pathway. Critical for predicting DDI magnitude. |
| Michaelis Constant | Km | µM | Substrate concentration at half Vmax. Determines enzyme saturation. |
| Maximal Reaction Velocity | Vmax | pmol/min/pmol enzyme | Intrinsic metabolic activity. |
| Hepatic Intrinsic Clearance | CLint | µL/min/mg protein | In vitro scaled intrinsic metabolic clearance. |
| Plasma Protein Binding | fu,p | Unitless | Fraction unbound in plasma. Impacts free drug concentration. |
| Blood-to-Plasma Ratio | B:P | Unitless | Partitioning of drug between blood cells and plasma. |
Table 2: Essential Perpetrator Drug Parameters for Enzyme Modulation
| Parameter | Symbol | Typical Units | Description & Relevance |
|---|---|---|---|
| Inhibitor Constant (Reversible) | Ki | µM | Concentration causing half-maximal inhibition. Used for static and dynamic models. |
| Inhibition Mechanism | - | - | Competitive, Non-competitive, Uncompetitive, Mixed. Informs model equations. |
| Maximum Inactivation Rate | kinact | min-1 | Maximal rate of enzyme inactivation (for time-dependent inhibitors). |
| Inactivator Concentration at half kinact | KI | µM | Concentration for half-maximal inactivation (for time-dependent inhibitors). |
| Induction EC50 | EC50 | µM | Concentration causing half-maximal enzyme induction. |
| Maximum Induction Effect | Emax | Fold-change | Maximal increase in enzyme activity or expression. |
2. Experimental Protocols for Parameter Generation
Protocol 1: Determination of Victim Drug fm and CLint Using Human Liver Microsomes (HLM) Objective: To quantify the intrinsic metabolic clearance and enzyme-specific fraction metabolized (fm) of a victim drug. Materials: See "Research Reagent Solutions" below. Procedure:
- Prepare incubation mixtures (final volume 100 µL) containing: 0.1 M phosphate buffer (pH 7.4), HLM (0.25-0.5 mg/mL), victim drug at a concentration << Km (typically 1 µM), and an NADPH-regenerating system.
- For reaction phenotyping, include selective chemical inhibitors (e.g., 1 µM ketoconazole for CYP3A4, 10 µM quinidine for CYP2D6) or use recombinant CYP isoforms individually.
- Pre-incubate mixtures at 37°C for 3 minutes. Initiate reactions by adding the NADPH-regenerating system.
- Terminate reactions at pre-determined time points (e.g., 0, 5, 10, 20, 30 min) by adding 100 µL of ice-cold acetonitrile containing an internal standard.
- Centrifuge at 4000×g for 10 minutes to pellet protein. Analyze supernatant using LC-MS/MS.
- Calculate in vitro CLint from the substrate depletion rate. Calculate fm,CYPi as the fraction of total CLint inhibited by a selective inhibitor or attributed to a specific recombinant CYP.
Protocol 2: Determination of Reversible Inhibition (Ki) for a Perpetrator Drug Objective: To characterize the potency of a perpetrator drug as a reversible enzyme inhibitor. Procedure:
- Prepare incubation mixtures with a probe substrate (e.g., midazolam for CYP3A4) at a concentration near its Km value.
- Include varying concentrations of the perpetrator inhibitor, spanning a range above and below the expected Ki.
- Follow steps 1 and 3-5 from Protocol 1.
- Determine the IC50 (concentration causing 50% inhibition of probe activity) by plotting % remaining activity vs. inhibitor concentration.
- Calculate Ki using the Cheng-Prusoff equation: Ki = IC50 / (1 + [S]/Km), where [S] is the probe substrate concentration.
Protocol 3: Assessment of Time-Dependent Inhibition (kinact, KI) Objective: To characterize mechanism-based or time-dependent enzyme inactivation. Procedure:
- Pre-incubation: Incubate HLM with perpetrator drug (multiple concentrations) and NADPH for a time course (e.g., 0, 5, 10, 20, 30 min).
- Dilution: Dilute the pre-incubation mixture 10-20 fold into a secondary incubation mixture containing a low concentration of probe substrate and NADPH.
- Secondary Reaction: Incubate for a short, fixed period (e.g., 5 min) to measure residual enzyme activity.
- Analysis: Determine the remaining enzyme activity at each pre-incubation time and inhibitor concentration. Fit the data to a nonlinear model to obtain kinact (the maximum inactivation rate) and KI (the inactivator concentration yielding half-maximal kinact).
3. Visualizing DDI Pathways and Workflows
Title: Perpetrator-Victim-CYP Interaction Pathways in DDI
Title: PBPK Modeling Workflow for Anti-Infective DDI Prediction
4. Research Reagent Solutions
| Item | Function in DDI Studies | Example/Supplier Note |
|---|---|---|
| Pooled Human Liver Microsomes (HLM) | Contains full complement of human CYP enzymes for metabolic stability, inhibition, and reaction phenotyping studies. | XenoTech, Corning Life Sciences. |
| Recombinant Human CYP Enzymes (rCYP) | Individual CYP isoforms for definitive reaction phenotyping and obtaining isoform-specific kinetic parameters. | BD Biosciences, Thermo Fisher. |
| CYP-Specific Probe Substrates & Inhibitors | Validated, selective compounds to measure activity of specific CYP enzymes (e.g., midazolam for CYP3A4). | Available as kits from vendors like Promega. |
| NADPH Regenerating System | Provides constant supply of NADPH, the essential cofactor for CYP-mediated reactions. | Often prepared from glucose-6-phosphate and dehydrogenase or purchased as solutions. |
| LC-MS/MS System | Gold standard for quantitative analysis of drugs and metabolites in complex biological matrices with high sensitivity and specificity. | Sciex, Thermo Fisher, Waters. |
| PBPK Software Platform | In silico environment for integrating in vitro data, building physiological models, and simulating DDIs. | Simcyp Simulator, GastroPlus, PK-Sim. |
Within the thesis on developing PBPK (Physiologically-Based Pharmacokinetic) models for predicting drug-drug interactions (DDIs) in anti-infective therapy, a critical advancement lies in the explicit incorporation of system-level physiological variability. The efficacy and toxicity of antimicrobial agents, and their interaction potential, are significantly modulated by patient-specific factors such as age, hepatic/renal impairment, and pharmacogenetics. This application note details protocols for integrating these covariates into PBPK models to enhance the predictive accuracy of DDI outcomes in diverse patient populations.
Quantitative Data on Key Physiological Variability Factors
Table 1: Age-Dependent Physiological Parameters Impacting Anti-infective PK
| Physiological Parameter | Neonate (0-1 mo) | Adult (20-50 yrs) | Elderly (≥65 yrs) | Primary Impacted Anti-infective Class |
|---|---|---|---|---|
| Glomerular Filtration Rate (mL/min/1.73m²) | ~20-40 | ~120 | ~60-80 | Aminoglycosides, β-lactams, Glycopeptides |
| Hepatic CYP3A4 Activity (% of adult) | 30-50% | 100% | 70-85% | Macrolides, Azole Antifungals, NNRTIs |
| Body Water (% total body weight) | 75% | 60% | 50-55% | Hydrophilic agents (e.g., Acyclovir) |
| Albumin (g/L) | 28-44 | 35-50 | 30-45 | Highly protein-bound drugs (e.g., Ceftriaxone) |
| Gastric pH | Elevated (pH ~6-8) | ~1.5-3.5 | Slightly Elevated | Azole antifungals (e.g., Itraconazole) |
Table 2: Impact of Organ Impairment on Drug Clearance Pathways
| Organ Impairment | Affected Clearance Pathway | Typical Reduction in Intrinsic Clearance | Example Anti-infective Requiring Dose Adjustment |
|---|---|---|---|
| Moderate Hepatic (Child-Pugh B) | CYP-mediated metabolism | 20-50% | Voriconazole, Efavirenz |
| Severe Hepatic (Child-Pugh C) | CYP-mediated metabolism & Biliary excretion | 50-80% | Rifampin, Erythromycin |
| Moderate Renal (eGFR 30-59) | Renal excretion | Proportional to eGFR reduction | Vancomycin, Penicillin G, Acyclovir |
| Severe Renal (eGFR <30) | Renal excretion | Proportional to eGFR reduction | Aminoglycosides, Polymyxins |
Table 3: Key Genetic Polymorphisms Affecting Anti-infective PK/DDI Risk
| Gene (Enzyme/Transporter) | Common Variant | Functional Consequence | Relevant Anti-infective & DDI Risk |
|---|---|---|---|
| CYP2C19 | *2, *3 (Loss-of-function) | Reduced enzyme activity | Voriconazole: Increased exposure, amplified DDI risk with CYP3A4 inhibitors. |
| CYP2B6 | 516G>T | Reduced enzyme activity | Efavirenz: Markedly increased exposure, potentiating CNS toxicity. |
| SLCO1B1 (OATP1B1) | 521T>C (Val174Ala) | Reduced hepatic uptake | Rifampin: Altered hepatic distribution, may modulate DDI magnitude with OATP substrates. |
| NAT2 | Slow acetylator alleles | Reduced acetylation rate | Isoniazid: Increased exposure and hepatotoxicity risk. |
Experimental Protocols for Data Generation & Model Parameterization
Protocol 3.1:In VitroDetermination of Fraction Metabolized (fm) by Specific CYP Enzymes Using Human Recombinant Enzymes
Objective: To quantify the fraction of an anti-infective drug metabolized by specific CYP isoforms (e.g., CYP3A4, CYP2C19) for accurate prediction of DDI magnitude in populations with genetic polymorphisms or organ impairment.
Materials (Research Reagent Solutions):
- Recombinant Human CYP Enzymes (Supersomes): Individual CYP isoforms (e.g., CYP3A4, 2C19, 2D6) co-expressed with human P450 reductase and cytochrome b5.
- NADPH Regenerating System: Provides a constant supply of NADPH for enzymatic reactions.
- LC-MS/MS System: For sensitive and specific quantification of parent drug and metabolites.
- Specific Chemical Inhibitors: e.g., Ketoconazole (CYP3A4), Ticlopidine (CYP2C19), Quinidine (CYP2D6) for reaction phenotyping verification.
- Pooled Human Liver Microsomes (HLM): Used as a comparator system.
Detailed Methodology:
- Incubation Setup: Prepare incubations containing the test anti-infective drug (at ~1 µM, near therapeutic Km), individual recombinant CYP enzymes (10-50 pmol/mL), and NADPH regenerating system in potassium phosphate buffer (pH 7.4). Run control incubations without NADPH.
- Time-Course Experiment: Terminate reactions at 0, 5, 10, 20, 30, and 45 minutes by transferring aliquots to ice-cold acetonitrile containing internal standard.
- Inhibition Confirmation: In parallel, perform incubations with HLM in the presence and absence of isoform-specific chemical inhibitors.
- Sample Analysis: Centrifuge to precipitate protein. Analyze supernatant via validated LC-MS/MS method to quantify depletion of parent drug.
- Data Analysis: Calculate initial depletion rates. The relative activity factor (RAF) for each recombinant enzyme may be applied to scale to HLM activity. The fraction metabolized (fmCYP) by a specific isoform is estimated from the relative depletion rate compared to total depletion in HLM.
Protocol 3.2: Population PK Study Design for Covariate Analysis
Objective: To collect clinical PK data across a diverse patient population to identify and quantify the impact of physiological covariates (age, organ function, genetics) on anti-infective PK parameters.
Materials:
- Validated Bioanalytical Assay: For drug and major metabolite quantification in plasma.
- Pharmacogenetic Testing Kit: TaqMan SNP Genotyping Assays for key polymorphisms (see Table 3).
- Electronic Data Capture (EDC) System: For recording precise demographics, clinical lab values (eGFR, Child-Pugh score), concomitant medications.
Detailed Methodology:
- Subject Recruitment: Enroll patients receiving the anti-infective drug of interest in a real-world clinical setting. Stratify recruitment to ensure representation across age groups, renal/hepatic function categories, and relevant ethnicities.
- Sparse Sampling: Employ an optimized, sparse sampling scheme (e.g., 2-4 samples per patient at pre-dose, 1-2 hr, 4-6 hr, and trough) to maximize information while minimizing patient burden.
- Covariate Measurement: Record age, weight, height, serum creatinine, albumin, bilirubin, INR, concomitant medications. Obtain blood sample for DNA isolation and genotyping.
- Modeling Analysis: Analyze data using nonlinear mixed-effects modeling (e.g., NONMEM). Develop a base structural PK model, then sequentially test inclusion of covariates (e.g., eGFR on clearance, weight on volume, genotype on metabolic clearance) using stepwise forward addition/backward elimination. Validate final model using visual predictive checks.
Visualization of Pathways and Workflows
Diagram Title: PBPK Model Integration of Physiological Variability
Diagram Title: Workflow for Building a Variability-Informed PBPK-DDI Model
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function/Application | Example Product/Catalog |
|---|---|---|
| Pooled Human Liver Microsomes (HLM) | In vitro system containing full complement of human drug-metabolizing enzymes for intrinsic clearance (CLint) and reaction phenotyping studies. | XenoTech H0610 (Mixed Gender) |
| Recombinant Human CYP Enzymes | Individual CYP isoforms for determining enzyme-specific kinetic parameters (Km, Vmax) and fraction metabolized (fm). | Corning Gentest Supersomes |
| Caco-2 Cell Line | Model for studying intestinal permeability and efflux transporter (e.g., P-gp) interactions relevant to oral anti-infective absorption and DDI. | ATCC HTB-37 |
| HEK293 Cells Overexpressing Transporters | Used to characterize substrate/inhibitor interactions with key hepatic/renal transporters (e.g., OATP1B1, OAT3, MATEs). | GenScript Transporter Assay Services |
| NADPH Regenerating System | Provides essential cofactor for CYP-mediated oxidative metabolism in microsomal incubations. | Promega V9510 |
| LC-MS/MS System with UPLC | Gold standard for sensitive, specific, and high-throughput quantification of drugs and metabolites in biological matrices for PK studies. | Waters ACQUITY UPLC & Xevo TQ-S micro |
| Population PK Modeling Software | Nonlinear mixed-effects modeling platform for covariate analysis and PK parameter estimation from clinical data. | NONMEM (ICON PLC), Monolix (Lixoft) |
| Whole-Body PBPK Simulation Platform | Software for building, simulating, and validating mechanistic PBPK models. | GastroPlus (Simulations Plus), PK-Sim (Open Systems Pharmacology) |
Within the context of a thesis on PBPK modeling for drug-drug interactions (DDI) in anti-infective therapy research, the selection of a robust software platform is critical. These tools enable the prediction of complex pharmacokinetic (PK) interactions, particularly vital when co-administering anti-infectives (e.g., HIV protease inhibitors, antimalarials, antifungals) with other medications. This application note provides a comparative overview of three leading platforms—Simcyp Simulator, GastroPlus, and PK-Sim/Open Systems Pharmacology Suite—detailing their application, protocols for DDI analysis, and essential research resources.
Table 1: Core Features and Applications in Anti-infective DDI Research
| Feature/Aspect | Simcyp Simulator | GastroPlus | PK-Sim / Open Systems Pharmacology |
|---|---|---|---|
| Primary Developer | Certara | Simulations Plus | Open Systems Pharmacology |
| Core Strengths | Population-based ADME, extensive DDI library, virtual populations. | Robust GI absorption (ACAT model), IVIVE, formulation modeling. | Open-source, modular, whole-body physiology integration. |
| Key Enzymes/Transporters | Full suite of CYPs, UGTs, and key transporters (P-gp, BCRP, OATPs, etc.). | Comprehensive CYP, UGT, and transporter networks. | Extensive list, customizable via open-source ontology. |
| Anti-infective Specific Libraries | Dedicated modules for antiretrovirals, antifungals, and antibiotics. | Built-in compound databases for common anti-infectives. | User-expandable compound database; community models available. |
| Typical DDI Prediction Accuracy (Reported Range) | 80-90% for CYP-mediated interactions. | 75-89% for mechanism-based inhibition. | Comparable accuracy, dependent on model parameterization. |
| Regulatory Use Citation | Frequently cited in US FDA and EMA submissions. | Supported in numerous regulatory filings. | Growing acceptance in regulatory submissions. |
Application Notes & Protocols
Protocol 1: Simulating a CYP3A4-Mediated DDI for an Antiretroviral
Objective: To predict the effect of a strong CYP3A4 inhibitor (e.g., Ketoconazole) on the PK of a new protease inhibitor.
Workflow:
- Compound File Preparation: Create a new drug compound file for the investigational anti-infective. Enter physicochemical properties (logP, pKa), blood-to-plasma ratio, and in vitro PK data (f~u~, CL~int~). Define the metabolic pathway, assigning a fraction metabolized by CYP3A4 (f~m,CYP3A4~) and entering the relevant K~m~ and V~max~/CL~int~ values from human liver microsomes.
- Population Selection: In the Simulator, select the "Simcyp Healthy Volunteer" population or a specific population (e.g., "HIV-Infected" if available). Set trial design (n=10 trials, 10 subjects/trial, appropriate dosing).
- DDI Scenario Setup: Navigate to the DDI module. Define the victim drug (new protease inhibitor) with its regimen. Define the perpetrator drug (Ketoconazole) from the built-in library. Set its regimen (e.g., 400 mg QD for 7 days).
- Simulation Execution & Analysis: Run the simulation. Outputs include geometric mean AUC and C~max~ ratios (DDI magnitude). Compare the predicted AUC ratio with the predefined clinical DDI risk threshold (e.g., ≥2-fold increase indicates positive interaction).
Title: Simcyp DDI Simulation Workflow for Anti-infectives
Protocol 2: Building a First-in-Human PBPK Model for an Antimalarial with GastroPlus
Objective: To develop and validate a PBPK model for a new antimalarial drug to support first-in-human (FIH) dose prediction and DDI risk assessment.
Workflow:
- Compound Profiling: In the
Compoundtab, input API properties (molecular weight, logP, pKa, solubility profile). In thePhysiology & PKtab, enter in vitro clearance (e.g., hepatocyte CL~int~), plasma protein binding data, and permeability (e.g., Caco-2, P~app~). - Model Building with IVIVE: Use the built-in IVIVE (in vitro to in vivo extrapolation) tools. The software will convert CL~int~ to human hepatic clearance using appropriate scaling factors and liver models.
- Model Optimization/Validation: If human PK data is available (even from a single dose), use the
Parameter Optimizationmodule to refine key parameters (e.g., effective permeability, fraction unbound) to match observed data. - DDI Prediction: Access the
DDImodule. Assign the antimalarial as a victim. Add a known CYP perpetrator (e.g., Rifampin for CYP induction). Simulate the co-administration scenario and evaluate changes in exposure.
Title: GastroPlus First-in-Human PBPK/DDI Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for In Vitro Data Generation for PBPK Input
| Research Reagent / Material | Function in PBPK Context | Key Provider Examples |
|---|---|---|
| Human Liver Microsomes (HLM) | Provide CYP enzyme activity for measuring intrinsic clearance (CL~int~) and inhibition constants (K~i~). | Corning Life Sciences, Thermo Fisher Scientific, XenoTech |
| Cryopreserved Human Hepatocytes | Used for measuring metabolic stability, identifying pathways, and assessing time-dependent inhibition (TDI). | BioIVT, Lonza, Thermo Fisher Scientific |
| Transfected Cell Systems (e.g., OATP-HEK293) | Express single human transporters to determine substrate specificity and kinetics (K~m~, V~max~) for transporter models. | Solvo Biotechnology, Corning Life Sciences |
| Caco-2 Cell Line | Standard model for assessing intestinal permeability, a critical input for absorption prediction in PBPK. | ATCC, Sigma-Aldrich |
| Human Plasma | Used to determine fraction unbound in plasma (f~u~), critical for accurate distribution and clearance predictions. | BioIVT, Sigma-Aldrich |
| Specific Chemical Inhibitors (e.g., Ketoconazole, Quinidine) | Used in reaction phenotyping experiments to identify the fraction of metabolism (f~m~) by specific CYP enzymes. | Sigma-Aldrich, Cayman Chemical |
Application Notes & Protocols
Thesis Context: This work forms a pivotal case study within a broader thesis on advancing Physiologically-Based Pharmacokinetic (PBPK) modeling frameworks to predict complex drug-drug interactions (DDIs) in anti-infective therapy. The goal is to enhance model-informed drug development (MIDD) for novel agents, ensuring safe and effective co-administration with standard therapies like rifampin.
1. Introduction & Rationale Rifampin is a potent inducer of cytochrome P450 3A4 (CYP3A4) and P-glycoprotein (P-gp). For a novel antiviral under development, predicting the magnitude of rifampin-dependent induction is critical to inform clinical DDI study design and potential dosing adjustments. This protocol details the in vitro to in vivo extrapolation (IVIVE) workflow integrated into a whole-body PBPK model to predict this interaction.
2. Key Experimental Data & Input Parameters Live search data indicates current standard practices for induction assessment, typically using human hepatocytes. Key quantitative parameters are summarized below.
Table 1: Novel Antiviral Compound Properties
| Parameter | Value | Source/Note |
|---|---|---|
| Molecular Weight | 450.2 g/mol | Calculated |
| logP | 3.1 | Predicted (ACD Labs) |
| fu, plasma | 0.15 (15%) | Human plasma protein binding assay |
| B:P Ratio | 0.8 | Blood cell partitioning assay |
| Primary Metabolizing Enzyme | CYP3A4 (>80%) | Reaction phenotyping (rCYP) |
| Contribution of CYP3A4 (fmCYP3A4) | 0.85 | Relative activity factor (RAF) approach |
Table 2: In Vitro Induction Parameters (Human Hepatocytes)
| Parameter | Value (Mean ± SD) | Experimental System |
|---|---|---|
| Rifampin EC₅₀ | 0.65 ± 0.21 µM | Cryopreserved HH, 48-72h incubation |
| Rifampin Emax | 12.5 ± 2.1-fold | CYP3A4 mRNA relative to vehicle |
| Novel Antiviral EC₅₀ | >30 µM (No significant induction) | Same as above (Negative control) |
| Novel Antiviral Emax | <2.0-fold | Confirms lack of self-induction |
3. Detailed Experimental Protocols
Protocol 3.1: CYP3A4 Induction Assay in Cryopreserved Human Hepatocytes Objective: To determine the concentration-dependent induction of CYP3A4 mRNA by rifampin and the novel antiviral. Materials: See "Scientist's Toolkit" below. Procedure:
- Thawing & Plating: Rapidly thaw cryopreserved human hepatocytes (3-donor pool) and plate in collagen-coated 96-well plates at a density of 0.7 x 10⁵ viable cells/well in incubation medium.
- Recovery: Incubate cells for 6-8 hours at 37°C, 5% CO₂.
- Dosing: Prepare serial dilutions of rifampin (0.1-50 µM) and the novel antiviral (0.3-100 µM) in treatment medium. Replace recovery medium with treatment medium containing test articles or vehicle (0.1% DMSO). Include positive control (rifampin at 10 µM).
- Incubation: Incubate cells for 48 hours, refreshing treatment medium at 24 hours.
- mRNA Quantification: Lyse cells and extract total RNA. Perform reverse transcription followed by quantitative real-time PCR (qRT-PCR) using TaqMan assays for CYP3A4 and the housekeeping gene GAPDH.
- Data Analysis: Calculate fold-induction over vehicle using the ΔΔCt method. Fit rifampin concentration-response data to a sigmoidal Emax model to derive EC₅₀ and Emax.
Protocol 3.2: PBPK Model Development & DDI Prediction Objective: To build and verify a PBPK model for rifampin, then use it to predict the effect on the novel antiviral's pharmacokinetics. Software: GastroPlus or PK-Sim. Procedure:
- Rifampin Model: Develop a full PBPK model for rifampin using published physicochemical, in vitro, and clinical PK data. Optimize and verify the model against independent clinical PK profiles (single and multiple doses).
- Novel Antiviral Model: Develop a minimal PBPK (mPBPK) model for the novel antiviral using data from Table 1 and Phase I single ascending dose (SAD) data.
- DDI Mechanistic Integration: Incorporate the induction parameters from Table 2. Use the rifampin model to simulate its unbound liver concentration-time profile. Link this to the induction model:
Induction = 1 + (Emax * C_u,liv^γ) / (EC₅₀^γ + C_u,liv^γ)to time-dependently increase CYP3A4 abundance and activity in the antiviral's clearance pathway. - Simulation: Simulate the plasma concentration-time profile of the novel antiviral following multiple dosing (e.g., 200 mg BID for 7 days) both alone and when co-administered with rifampin (600 mg QD for 7 days).
- Output: Predict the change in key exposure metrics: Area Under the Curve (AUC) and maximum concentration (Cmax).
4. Visualization
Diagram 1 Title: PBPK-IVIVE Workflow for Induction DDI Prediction
Diagram 2 Title: Rifampin's PXR-Mediated Induction Mechanism
5. The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Protocol 3.1 | Example Vendor/Product |
|---|---|---|
| Cryopreserved Human Hepatocytes (3-donor pool) | Biologically relevant system expressing functional nuclear receptors (PXR) and drug-metabolizing enzymes. | BioIVT (Hu4123), Lonza |
| Collagen-Coated 96-Well Plates | Provides extracellular matrix for hepatocyte attachment and maintenance of polarized morphology. | Corning (BioCoat) |
| Hepatocyte Incubation/Treatment Medium | Serum-free, hormonally defined medium optimized for hepatocyte function and induction response. | Thermo Fisher (Williams' E Medium) |
| Rifampin (Reference Standard) | Potent PXR agonist used as a positive control and for calibration of the induction response. | Sigma-Aldrich (R3501) |
| TaqMan Gene Expression Assays | Fluorogenic probes for specific, sensitive quantification of CYP3A4 and housekeeping gene mRNA. | Thermo Fisher (Hs00604506_m1 for CYP3A4) |
| RNA Isolation Kit | Rapid purification of high-quality total RNA from small numbers of plated hepatocytes. | Qiagen (RNeasy 96 Kit) |
| Data Analysis Software | For non-linear regression to fit induction EC₅₀/Emax models (e.g., GraphPad Prism). | GraphPad Prism |
Overcoming Challenges: Troubleshooting and Refining PBPK DDI Models
This document, framed within a broader thesis on Physiologically-Based Pharmacokinetic (PBPK) modeling for drug-drug interactions (DDIs) in anti-infective therapy research, details critical methodological pitfalls. The reliable prediction of DDIs is paramount for anti-infective agents (e.g., protease inhibitors, macrolides, azoles), which are often perpetrators or victims of cytochrome P450 (CYP)- and transporter-mediated interactions. This application note provides structured protocols and data to aid researchers in navigating parameter sensitivity, model misspecification, and overfitting.
Key Pitfalls: Definitions and Quantitative Impact
Parameter Sensitivity
The disproportionate influence of a specific model input on the output. In PBPK-DDI, highly sensitive parameters demand precise estimation.
Table 1: High-Sensitivity Parameters in Anti-Infective PBPK Models
| Parameter | Typical Range | Impact on AUC Ratio (Victim Drug) | Key Enzyme/Transporter |
|---|---|---|---|
| Fraction unbound in plasma (fu) | 0.01 - 0.2 | ± 40-60% for high-extraction drugs | N/A |
| Intrinsic clearance (CLint) | 1 - 500 µL/min/mg | Direct determinant of baseline exposure | CYP3A4, CYP2C19 |
| Inhibition constant (Ki) | 0.01 - 10 µM | ± 70-90% for strong inhibitors | CYP3A4 (e.g., ritonavir) |
| Biliary clearance (CLbile) | 0.1 - 50 L/h | ± 30-50% for hepatically cleared drugs | P-gp, BCRP, MRP2 |
Model Misspecification
An error in the fundamental model structure, such as omitting a relevant metabolic pathway or transporter interaction.
Table 2: Common Misspecifications in Anti-Infective DDI Models
| Misspecification | Consequence | Example in Anti-Infectives |
|---|---|---|
| Omitting gut metabolism | Underprediction of first-pass effect, mispredicted perpetrator strength | Macrolides (e.g., clarithromycin) impacting gut CYP3A. |
| Assuming only competitive inhibition | Misprediction of time-dependent DDI dynamics | Time-dependent inhibition by protease inhibitors (e.g., lopinavir/ritonavir). |
| Ignoring transporter interplay (e.g., hepatic uptake + metabolism) | Incorrect prediction of hepatic concentration and DDI magnitude | Rifampicin's OATP-mediated uptake affecting its CYP-mediated metabolism. |
| Using healthy volunteer physiology for special populations | Inaccurate dose recommendations | DDI in patients with hepatic impairment (e.g., voriconazole). |
Overfitting
The model describes the calibration dataset with high precision but fails to predict new data reliably, often due to excessive parameter optimization or unnecessary complexity.
Table 3: Indicators of Overfitting in PBPK Model Development
| Indicator | Acceptable Threshold | Overfitting Warning Sign |
|---|---|---|
| Objective Function Value (OFV) reduction per added parameter | > 3.84 (χ², p<0.05) | < 1.0 |
| Normalized Prediction Distribution Error (NPDE) | Mean ≈ 0, Variance ≈ 1 | Significant deviation from theoretical N(0,1) |
| Visual Predictive Check (VPC) | 90% of observed data within 90% prediction interval | >95% of data within interval for calibration set, but poor in validation. |
| Number of optimized parameters relative to data points | < 1:5 (Parameters:Observations) | > 1:2 |
Experimental Protocols
Protocol 1: Global Sensitivity Analysis (GSA) for Parameter Prioritization
Objective: To systematically identify and rank parameters influencing DDI AUC predictions. Materials: PBPK software (e.g., Simcyp, GastroPlus, PK-Sim), in vitro inhibition/induction data. Procedure:
- Define Model & Output: Establish a verified base PBPK model for both perpetrator (e.g., clarithromycin) and victim drug (e.g., midazolam). Define the output of interest (e.g., AUC ratio of victim with/without perpetrator).
- Set Parameter Ranges: Assign plausible physiological (e.g., liver blood flow) and drug-specific (e.g., Ki, fu) parameter distributions (e.g., uniform ± 30% of baseline).
- Sampling: Use a Latin Hypercube Sampling (LHS) scheme to generate 1000-5000 parameter sets across the defined multidimensional space.
- Simulation & Analysis: Run the DDI simulation for each parameter set. Perform a variance-based sensitivity analysis (e.g., Sobol method) to compute total-order sensitivity indices (STi) for each input parameter.
- Interpretation: Parameters with STi > 0.1 are considered highly sensitive and require rigorous in vitro/in vivo verification.
Protocol 2: Model Qualification to Detect Misspecification
Objective: To test the structural adequacy of a PBPK-DDI model using independent data. Materials: Clinical DDI studies not used for model development (different dosing regimens, populations, or co-administered drugs). Procedure:
- Split Data: Reserve 20-30% of available clinical DDI studies for qualification. Do not use this data for any model calibration.
- Predict & Compare: Using the fully calibrated model, predict the pharmacokinetic (PK) profiles and AUC ratios for the qualification studies.
- Apply Validation Metrics: Calculate the average fold error (AFE) and absolute average fold error (AAFE). Apply the 2-fold acceptance criterion: AAFE ≤ 2.0 indicates successful qualification.
- Root Cause Analysis: If criteria fail (e.g., AAFE > 2.0), systematically test alternative model structures (e.g., add transporter mediation, different inhibition mechanisms) on the calibration set.
Protocol 3: Preventing Overfitting via Cross-Validation
Objective: To ensure model generalizability and avoid excessive parameterization. Materials: A comprehensive dataset of observed PK profiles from clinical DDI studies. Procedure:
- Data Partitioning: Randomly partition the clinical dataset into k folds (e.g., k=5).
- Iterative Training/Validation: For i = 1 to k:
- Designate fold i as the temporary validation set.
- Calibrate (optimize) the model parameters using the remaining k-1 folds.
- Predict the PK in fold i and calculate prediction errors.
- Aggregate Performance: Compute the overall prediction error (e.g., RMSE) across all k iterations. This cross-validated error is a robust measure of predictive performance.
- Model Simplification: If cross-validated error is significantly larger than calibration error, simplify the model (e.g., fix uncertain parameters to literature values, remove unnecessary compartments) and repeat the process.
Visualizations
Title: PBPK-DDI Model Robustness Workflow
Title: Hepatic CYP Inhibition DDI Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Tools for PBPK-DDI Model Development in Anti-Infectives
| Item/Reagent | Function in DDI Research | Example/Supplier |
|---|---|---|
| Recombinant CYP Enzymes | Determine enzyme-specific intrinsic clearance (CLint) and inhibition constants (Ki). | Baculovirus-insect cell expressed CYPs (e.g., Supersomes, Corning). |
| Transfected Cell Systems | Assess transporter-mediated uptake/efflux (e.g., OATP1B1, P-gp). | MDCKII or HEK293 cells overexpressing human transporters. |
| Human Liver Microsomes (HLM) & Hepatocytes | Measure metabolic stability, reaction phenotyping, and time-dependent inhibition (TDI). | Pooled HLM (e.g., XenoTech, BioIVT); cryopreserved hepatocytes. |
| LC-MS/MS System | Quantify drug and metabolite concentrations in in vitro assays and clinical samples. | Triple quadrupole systems (e.g., Sciex, Waters, Thermo). |
| PBPK Software Platform | Integrate in vitro data, simulate PK and DDI in virtual populations. | Simcyp Simulator, GastroPlus, PK-Sim. |
| Clinical DDI Database | For model calibration and qualification. | University of Washington Drug Interaction Database, published literature. |
Within the context of PBPK modeling for drug-drug interactions (DDIs) in anti-infective therapy, a critical challenge is the presence of unclear parameters, such as unbound fraction in tissues, precise enzyme/transporter kinetics in pathological states, and intracellular concentrations. Robust IVIVE strategies are essential to bridge these gaps and build predictive models for complex therapeutic regimens.
Key Data Gaps and Quantitative Summaries
Table 1: Common Unclear Parameters in Anti-Infective PBPK-DDI Modeling
| Parameter | Typical In Vitro Source | Major Source of Uncertainty for IVIVE | Potential Impact on DDI Prediction |
|---|---|---|---|
| Fraction Unbound in Tissue (fut) | Plasma protein binding assays, tissue homogenate binding | Scaling from homogenate to cellular partitioning; disease state alterations. | Under/over-prediction of tissue substrate concentration & perpetrator potency. |
| Hepatic Intrinsic Clearance (CLint) | Microsomes, hepatocytes (human) | Differences in isoform activity between systems; impact of infection/inflammation. | Misestimation of metabolic drug clearance and magnitude of enzyme-mediated DDIs. |
| Transporter Kinetic Parameters (Km, Vmax) | Overexpression cell systems (e.g., HEK293, MDCK) | Scaling factor for expression level difference; in vivo relevance of assay conditions. | Incorrect prediction of transporter-mediated uptake/efflux and associated DDIs. |
| Inhibitory Constant (Ki) | Recombinant enzyme or cell-based inhibition assays | Discrepancy between initial rate vs. end-point assays; mechanism of inhibition. | Error in predicting the inhibition potential of a perpetrator anti-infective agent. |
| Intracellular Concentration | Calculated from permeability assays | Active transport components; subcellular compartmentalization (e.g., lysosomes). | Poor prediction of efficacy for intracellular pathogens and associated DDIs. |
Table 2: Strategies for Addressing Parameter Uncertainties
| Strategy | Description | Application Example |
|---|---|---|
| Proteomics-informed Scaling | Using quantitative proteomic data for enzyme/transporter abundances to refine scaling factors. | Scaling hepatocyte CLint using disease-specific CYP3A4 abundance from liver biopsies. |
| Mechanistic Tissue Composition | Modeling tissue partitioning using composition-based models (e.g., Rodgers & Rowland). | Estimating fut for a lipophilic antifungal in lung tissue for pulmonary aspergillosis. |
| Transgenic Animal Data | Using humanized animal models to provide integrated in vivo parameters for human proteins. | Estimating in vivo Km for hepatic OATP1B1 using humanized OATP1B1 mouse data. |
| Sensitive Clinical DDI Data | Leveraging sparse clinical DDI data to inversely estimate key uncertain parameters via PBPK. | Optimizing Ki and fu for a new protease inhibitor using a small clinical DDI study with midazolam. |
| Bayesian Population PBPK | Using prior distributions for parameters and updating with available data to quantify uncertainty. | Characterizing the population variability in renal OCT2 activity in patients with multidrug-resistant TB. |
Experimental Protocols for Parameter Elucidation
Protocol 1: Determination of Mechanistic Fraction Unbound in Tissue (fut)
Objective: To experimentally determine fut for a drug in a specific tissue (e.g., liver, lung) to inform PBPK model tissue partitioning. Materials: Tissue of interest (human/animal), phosphate-buffered saline (PBS), rapid equilibrium dialysis (RED) device, test compound, LC-MS/MS system. Methodology:
- Homogenize tissue in PBS (1:4 w/v) using a mechanical homogenizer on ice.
- Spike the tissue homogenate with the test compound at a therapeutic relevant concentration (e.g., 1 µM).
- Load the spiked homogenate into the sample chamber of a RED insert. Load PBS into the buffer chamber.
- Incubate at 37°C with gentle agitation for 6-8 hours to reach equilibrium (optimize time preliminarily).
- Post-incubation, quantify compound concentration in both homogenate and buffer chambers using a validated LC-MS/MS method.
- Calculate fut = (Concentration in Buffer) / (Concentration in Homogenate). Correct for non-specific binding to the dialysis device using control experiments.
Protocol 2: Proteomics-Informed Scaling of Transporter Activity
Objective: To scale in vitro transporter Vmax from overexpression systems to physiological levels using quantitative proteomics. Materials: Overexpression cell system (e.g., HEK293-OATP1B1), cryopreserved human hepatocytes, mass spectrometry-compatible lysis buffer, trypsin, LC-MS/MS with data-independent acquisition (DIA). Methodology:
- Generate In Vitro Kinetic Data: Conduct uptake assays in HEK293-OATP1B1 cells to determine Vmax_app(in vitro) and Km for a probe substrate (e.g., estradiol-17β-D-glucuronide).
- Quantify Transporter Abundance: a. Lyse HEK293-OATP1B1 cells and pooled human hepatocyte samples (n≥5 donors) in RIPA buffer. b. Digest proteins with trypsin and analyze peptides using DIA proteomics. c. Quantify OATP1B1 abundance (fmol/µg total protein) using isotopically labeled peptide standards.
- Calculate Relative Expression Factor (REF): REF = [OATP1B1] in hepatocytes / [OATP1B1] in HEK293-OATP1B1.
- Scale Vmax: Vmaxpred(in vivo) = Vmaxapp(in vitro) × REF. Incorporate this scaled Vmax into the liver PBPK model compartment.
Visualization of Strategies and Workflows
Title: IVIVE Strategy Selection for Unclear PBPK Parameters
Title: Proteomics-Informed Scaling of Vmax Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for IVIVE Parameter Elucidation Experiments
| Item | Function in IVIVE Context | Example Product/Catalog |
|---|---|---|
| Cryopreserved Human Hepatocytes | Gold-standard cell system for measuring intrinsic hepatic clearance and metabolism-mediated DDIs. | BioIVT Human Hepatocytes, Corning Gentest Hepatocytes. |
| Transporter-Overexpressing Cell Lines | Isolate and quantify the kinetics of specific uptake (e.g., OATPs) or efflux (e.g., P-gp) transporters. | Solvo Biotechnology transfected cell lines, Thermo Fisher Flp-In TREx systems. |
| Rapid Equilibrium Dialysis (RED) Device | Determine fraction unbound in plasma or tissue homogenate for partitioning calculations. | Thermo Fisher Scientific RED Plate. |
| Isotopically Labeled Peptide Standards (SIS) | Absolute quantification of enzyme/transporter protein abundance via LC-MS/MS proteomics. | JPT Peptide Technologies SpikeTides, Thermo Fisher Scientific Pierce Retention Time Calibration Kit. |
| PBPK Modeling Software with Optimization | Platform to integrate in vitro data, apply scaling, and calibrate models using clinical data. | Certara Simcyp Simulator, Bayer PK-Sim, Open-Source mrgsolve (R). |
| Recombinant Human Cytochrome P450 Enzymes | Determine isoform-specific metabolic clearance and inhibition constants (Ki). | Corning Gentest Supersomes, Cypex Bactosomes. |
Within the broader thesis on PBPK modeling for drug-drug interactions (DDIs) in anti-infective therapy, a critical challenge is extrapolating predictions to special populations. Variability in renal/hepatic function and pediatric physiology profoundly alters pharmacokinetics (PK), complicating DDI risk assessment. Optimizing PBPK models for these populations is essential to predict exposure and DDI magnitude accurately, enabling dose adjustment strategies and informing clinical study design without exposing vulnerable patients to unnecessary risk.
Application Notes: Key Physiological Variability & Model Parameters
Table 1: Key System-Dependent Parameters for Special Population PBPK Modeling
| Physiological Parameter | Healthy Adult (Reference) | Renal Impairment (Severe) | Hepatic Impairment (Child-Pugh C) | Pediatrics (2-6 years) |
|---|---|---|---|---|
| Glomerular Filtration Rate (GFR) | 120 mL/min | 15-29 mL/min (Stage 4) | Largely preserved | Highly age-dependent; ~60-120 mL/min/1.73m² |
| Hepatic CYP3A4 Abundance | 100% | Mildly reduced (≈80-100%) | Significantly reduced (≈30-50%) | Maturation from ≈30% (1 yr) to ≈100% (adult) |
| Plasma Albumin | 4.0-4.5 g/dL | Often reduced (≈3.0-3.5 g/dL) | Significantly reduced (≈2.5-3.0 g/dL) | Slightly reduced (≈3.5-4.0 g/dL) |
| Hematocrit | 45% | Often reduced (anemia) | May be reduced | Age-dependent (≈35-39%) |
| Body Composition (Fat %) | Reference | Variable | May be altered (e.g., ascites) | Higher than adults, varies with age |
| Key Modeling Approach | Baseline Model | Scale renal clearance via GFR; adjust albumin if needed. | Adjust hepatic metabolic clearance (enzyme abundance/function), plasma protein binding. | Implement age-dependent functions for organ size, blood flows, enzyme maturation, GFR. |
Table 2: Impact on Anti-Infective PK and Exemplar Drugs
| Population | Primary PK Alteration | DDI Risk Implication | Exemplar Anti-Infectives |
|---|---|---|---|
| Renal Impairment | Increased exposure of renally cleared drugs; potential accumulation. | Altered victim drug clearance can modulate DDI magnitude (e.g., with CYP inhibitors). | Vancomycin, Aminoglycosides, Cidofovir, Remdesivir (metabolite GS-441524). |
| Hepatic Impairment | Increased exposure of hepatically cleared drugs; altered protein binding. | Reduced metabolic capacity can diminish DDI magnitude for enzyme-mediated interactions. | Voriconazole, Erythromycin, Rifampin, Simeprevir. |
| Pediatrics | Variable clearance (maturation), different volume of distribution. | DDI magnitude may differ from adults due to evolving enzyme/transporter activity. | Most drugs require careful extrapolation. |
Detailed Experimental Protocols
Protocol 1:In VitroDetermination of Fraction Metabolized (fm) for Hepatic Impairment Scaling
Objective: To quantify the fraction of drug metabolized by specific CYP enzymes using human liver microsomes (HLM) and chemical inhibitors, informing clearance scaling in hepatic impairment models. Materials: See "Scientist's Toolkit" below. Procedure:
- Incubation Setup: Prepare duplicate incubation mixtures containing: 0.1 M phosphate buffer (pH 7.4), 1 mM NADPH, HLM (0.5 mg/mL), and the anti-infective drug at a concentration near Km. Pre-incubate at 37°C for 5 min.
- Inhibitor Conditions: Include the following separate conditions:
- Negative Control: No NADPH.
- Positive Control: No inhibitor.
- Test Conditions: With selective chemical inhibitors (e.g., 1 µM Ketoconazole for CYP3A4, 10 µM Quinidine for CYP2D6).
- Reaction Initiation & Termination: Start reactions by adding NADPH. Incubate for a predetermined linear time (e.g., 20 min). Terminate by adding 2 volumes of acetonitrile with internal standard.
- Sample Analysis: Vortex, centrifuge (3000xg, 15 min), and analyze supernatant using LC-MS/MS to quantify parent drug depletion or metabolite formation.
- Data Analysis: Calculate reaction velocity (V) for each condition. Determine fraction metabolized by a specific pathway (fmCYPi) as: fmCYPi = 1 - (V with inhibitor / V positive control).
Protocol 2: Population PK (PopPK) Analysis to Inform Pediatric PBPK Prior Knowledge
Objective: To analyze sparse clinical PK data from pediatric trials to estimate key parameters (e.g., clearance maturation) for PBPK model verification. Materials: Pediatric PK dataset, NONMEM or Monolix software. Procedure:
- Structural Model Development: Start with a published adult PBPK/compartmental model. Implement a standard allometric function (e.g., (WT/70)^0.75) for clearance (CL) and volume (V) scaling.
- Covariate Model Building: Test maturation functions on clearance. For example, incorporate a sigmoidal maturation model: CL = CL_std * (WT/70)^0.75 * [PMA^HILL / (TM50^HILL + PMA^HILL)], where PMA is post-menstrual age and TM50 is maturation half-life.
- Model Estimation: Fit the model to pediatric data using nonlinear mixed-effects modeling. Estimate population parameters (typical CL, V, TM50, HILL) and inter-individual variability.
- Model Evaluation: Use diagnostic plots (observed vs. predicted, conditional weighted residuals) and visual predictive checks.
- Output for PBPK: The estimated TM50 and HILL for clearance are directly used to refine the physiological maturation function in the pediatric PBPK model.
Visualization of Workflows
Special Population PBPK Model Development Workflow
DDI Mechanistic Basis in Special Populations
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Key Protocols
| Item / Reagent | Supplier Examples | Function in Protocol |
|---|---|---|
| Human Liver Microsomes (HLM) | Corning Life Sciences, XenoTech LLC, BioIVT | Source of human drug-metabolizing enzymes for in vitro clearance and fm determination. |
| Recombinant CYP Enzymes | Corning Life Sciences, Cypex Ltd. | Isolated human CYP isoforms for reaction phenotyping and specific activity assessment. |
| Selective Chemical Inhibitors | Sigma-Aldrich, Cayman Chemical | To selectively inhibit specific CYP enzymes (e.g., Ketoconazole for CYP3A4) in HLM experiments. |
| NADPH Regenerating System | Promega Corporation, Corning Life Sciences | Provides constant supply of NADPH cofactor for oxidative metabolic reactions in microsomal incubations. |
| LC-MS/MS System | Sciex, Agilent, Waters, Thermo Fisher Scientific | Quantification of drug and metabolite concentrations in in vitro samples and biological fluids with high sensitivity. |
| Population PK Modeling Software | NONMEM (ICON plc), Monolix (Lixoft), Phoenix NLME (Certara) | For statistical analysis of sparse clinical PK data to estimate population parameters and covariate effects. |
| Whole PBPK Modeling Platform | GastroPlus (Simulations Plus), PK-Sim (Open Systems Pharmacology), Simcyp Simulator (Certara) | Integrates system, drug, and trial data to simulate PK and DDIs in virtual populations. |
Application Notes
Within the framework of PBPK modeling for drug-drug interactions (DDIs) in anti-infective therapy, model qualification is a critical step to establish predictive credibility prior to application in regulatory or clinical decision-making. Internal verification ensures the model's mathematical and logical integrity, while diagnostic evaluations assess its ability to recapitulate observed clinical DDI data. This process is fundamental for models intended to explore complex interactions between anti-infectives (e.g., azole antifungals, macrolides, rifamycins) and concomitant medications, which are common in treating co-infections or comorbidities.
Core Principles of Internal Verification
- Code Verification: Confirming the model software implementation is error-free and performs as designed.
- Mass Balance: Ensuring the total mass of the drug is conserved across all compartments over time.
- Sensitivity Analysis (Local/Global): Identifying and ranking model parameters (e.g., CYP enzyme Ki, fraction unbound, tissue permeability) that most significantly influence key DDI predictions (e.g., AUC ratio).
Key Diagnostic Metrics for DDI PBPK Models
Diagnostics focus on comparing model-simulated pharmacokinetic (PK) profiles and DDI magnitudes against clinically observed data. Acceptance criteria are often guided by regulatory expectations (e.g., FDA, EMA).
Table 1: Primary Diagnostic Metrics for Anti-infective DDI PBPK Models
| Metric | Description | Typical Qualification Criteria (for Anti-infectives) |
|---|---|---|
| AUC Ratio (AUCR) | Ratio of victim drug area under the curve with/without perpetrator. | Predicted vs. observed AUCR within 1.25-fold (or 0.8-1.25). |
| Cmax Ratio | Ratio of victim drug maximum concentration with/without perpetrator. | Predicted vs. observed Cmax ratio within 1.25-fold. |
| Visual Predictive Check (VPC) | Overlay of observed data percentiles with simulated prediction intervals. | ≥90% of observed data points fall within the 90% prediction interval. |
| Geometric Mean Fold Error (GMFE) | Measure of average fold error of predicted PK parameters. | GMFE for AUCR and Cmax ratio ≤ 1.25 (or ≤ 1.5 for more complex interactions). |
Table 2: Example Diagnostic Outcomes for a Rifampin-Midazolam DDI Model
| Perpetrator (Dose) | Victim (Dose) | Observed AUCR | Predicted AUCR (Mean ± SD) | GMFE (AUCR) | Qualification Status |
|---|---|---|---|---|---|
| Rifampin (600 mg QD) | Midazolam (2 mg IV) | 0.23 | 0.27 ± 0.05 | 1.15 | Pass |
| Rifampin (600 mg QD) | Midazolam (5 mg PO) | 0.10 | 0.12 ± 0.03 | 1.18 | Pass |
Experimental Protocols
Protocol 1: Internal Verification via Mass Balance Analysis
Objective: To verify that the total administered drug mass is accounted for in the PBPK system throughout the simulation.
Materials: Qualified PBPK software platform (e.g., GastroPlus, Simcyp Simulator, PK-Sim).
Procedure:
- Model Setup: Develop a base PBPK model for the anti-infective drug (e.g., voriconazole).
- Dosing Simulation: Simulate a single intravenous dose to eliminate absorption variability.
- Mass Output Configuration: Configure the software to output the cumulative mass in each compartment (plasma, tissues, liver, kidneys) and cumulative mass eliminated (via urine, feces, metabolism) at each time point.
- Calculation: For every simulated time point (t), calculate:
Total Mass(t) = Σ(Mass in all compartments) + Σ(Cumulative mass eliminated) - Verification: Plot Total Mass(t) over the simulation period. The result should be a horizontal line equal to the administered dose. Deviation >0.1% of dose indicates a mass balance error requiring model correction.
Protocol 2: Diagnostic Evaluation Using Clinical DDI Studies
Objective: To qualify the model's predictive performance for CYP3A4-mediated DDI between a perpetrator anti-infective (e.g., clarithromycin) and a sensitive victim drug (e.g., simvastatin).
Materials: PBPK software; digitized or published clinical PK data for victim drug alone and in combination; virtual population matching clinical study demographics.
Procedure:
- Base Model Qualification: Independently qualify the victim (simvastatin) and perpetrator (clarithromycin) PBPK models using single-agent PK data. Ensure predicted PK parameters (AUC, Cmax, CL) fall within 1.5-fold of observed values.
- DDI Mechanism Integration: Incorporate the appropriate interaction mechanism (e.g., competitive inhibition of CYP3A4 by clarithromycin and its metabolite). Input the in vitro-derived Ki value.
- Virtual Trial Replication: Design a virtual trial that mirrors the design of the clinical DDI study (dosing regimen, subject number, demographics, fasting/fed state).
- Simulation Execution: Run n=10 trials (≥100 virtual subjects each) to account for population variability.
- Diagnostic Analysis:
- Calculate the predicted geometric mean AUCR and Cmax ratio from the virtual trials.
- Compare to the observed clinical geometric mean ratio.
- Calculate the GMFE for both AUCR and Cmax.
- Generate a Visual Predictive Check (VPC) plot overlaying observed victim drug concentration-time profiles (with and without perpetrator) with the 5th, 50th, and 95th percentiles of the simulated profiles.
- Qualification Judgment: The model is considered qualified for this DDI scenario if the GMFE ≤ 1.25 and the VPC shows adequate concordance.
Visualizations
PBPK Model Qualification Workflow
CYP3A4 Inhibition DDI Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for PBPK Model Qualification in Anti-infective DDI
| Item / Solution | Function in Qualification | Example/Note |
|---|---|---|
| PBPK/PD Modeling Software | Platform for building, simulating, and verifying models. | GastroPlus, Simcyp Simulator, PK-Sim, MATLAB/SimBiology. |
| In Vitro Ki/IC50 Data | Quantitative measure of enzyme inhibition/induction potency for the perpetrator drug. | Generated from human liver microsomes or recombinant CYP enzymes. Critical input. |
| Digitized Clinical PK Data | Observed data used for diagnostic comparisons. | Extracted from literature using tools like WebPlotDigitizer; forms the gold standard. |
| Virtual Population Database | Genotypically/phenotypically diverse virtual subjects for trial simulation. | Simcyp's Virtual Populations, FDA's "Virtual Citizen." Must match clinical cohort. |
| Sensitivity Analysis Tool | Identifies parameters most influential on DDI predictions. | Built-in tools (e.g., Sobol, Morris) within PBPK platforms or standalone (R, SAAM II). |
| Visual Predictive Check (VPC) Script | Generates diagnostic plots comparing simulated and observed data percentiles. | Often requires custom coding in R or Python (using ggplot2, Matplotlib). |
Within the thesis on "PBPK Modeling for Drug-Drug Interactions in Anti-Infective Therapy Research," the development of robust models is critical. Anti-infectives are prone to complex DDIs due to enzyme/transporter induction/inhibition, protein binding displacement, and altered organ function in infected states. A priori PBPK models, built from in vitro and preclinical data, require calibration and validation with human data. This application note details the protocol for the iterative refinement of anti-infective PBPK models using early clinical pharmacokinetic data (e.g., from Phase I single ascending dose/multiple ascending dose studies) to enhance predictive accuracy for DDI simulations.
Protocol: Iterative Refinement Workflow
Objective: To systematically integrate early clinical PK data into a prior PBPK model, identify sensitive parameters, optimize the model, and validate its predictive performance for subsequent DDI simulations.
Materials & Software:
- Prior PBPK model file (e.g., *.pkml, *.model)
- Clinical PK dataset (Phase I SAD/MAD)
- PBPK software platform (e.g., PK-Sim, Simcyp Simulator, GastroPlus)
- Statistical software (e.g., R, Phoenix WinNonlin)
Procedure:
Step 1: Clinical Data Preparation & Model Configuration.
- Compile Phase I clinical PK data (plasma concentration-time profiles) for the anti-infective of interest. Include demographic data (weight, age, sex, genotype if available).
- In the PBPK software, replicate the clinical study design within a virtual population matching the trial's demographics and size (e.g., n=10 per dose group).
- Input the prior compound model (with defined absorption, distribution, metabolism, and excretion—ADME—parameters).
- Run an initial simulation and visually compare the simulated PK profiles with the observed clinical data.
Step 2: Sensitivity Analysis & Parameter Identification.
- Conduct a local or global sensitivity analysis on key ADME parameters (e.g., intrinsic clearance, fraction unbound, permeability, etc.) around the observed clinical PK metrics (AUC, C~max~, t~1/2~).
- Rank parameters by their normalized sensitivity coefficient. Identify the 3-5 parameters to which the model output is most sensitive (see Table 1).
Step 3: Model Optimization (Informing & Refining).
- Using the clinical PK data as the reference dataset, define objective functions (e.g., sum of squared errors, log-likelihood).
- Apply an optimization algorithm (e.g., Monte Carlo, particle swarm) to systematically adjust the a priori values of the sensitive parameters identified in Step 2 within physiologically plausible bounds.
- The goal is to minimize the discrepancy between simulated and observed PK profiles. The optimized parameter set now constitutes the informed PBPK model.
Step 4: Internal Validation & DDI Simulation.
- Validate the informed model against a separate subset of the clinical data not used in optimization (e.g., a different dose level or fed/fast state).
- If validation is successful (e.g., prediction within 1.5-2.0 fold), proceed to DDI simulation. Set up a virtual DDI study using the informed model for the anti-infective (as victim or perpetrator) and a validated model for the interacting drug (e.g., rifampin, itraconazole).
- Simulate the DDI and predict the geometric mean fold-change in AUC and C~max~.
Step 5: Comparison & Decision Making.
- Compare the DDI predictions from the informed model against those from the prior model.
- Assess the impact of refinement on the DDI risk assessment. Determine if the predicted DDI magnitude warrants clinical DDI study design modification or labeling considerations.
Data Presentation
Table 1: Example Output from Sensitivity Analysis for a Hypothetical Antifungal Drug
| Parameter | Description | Normalized Sensitivity Coefficient (AUC) | Rank | Physiological Plausible Range |
|---|---|---|---|---|
| CL_int | Hepatic intrinsic clearance | 1.85 | 1 | 5 - 50 µL/min/million cells |
| F_a | Fraction absorbed | 0.92 | 2 | 0.5 - 1.0 |
| Kpscalar | Tissue-plasma partition coefficient scalar | 0.45 | 3 | 0.3 - 3.0 |
| f_u | Fraction unbound in plasma | 0.38 | 4 | 0.01 - 0.15 |
| P_app | Apparent permeability | 0.12 | 5 | 1 - 20 x 10^-6 cm/s |
Table 2: Model Performance Before and After Iterative Refinement
| Metric | Prior Model Prediction | Informed Model Prediction | Observed Clinical Data (Mean) | Fold-Error (Prior) | Fold-Error (Informed) |
|---|---|---|---|---|---|
| AUC~0-24~ (mg·h/L) | 45.2 | 58.7 | 55.1 | 0.82 | 1.07 |
| C~max~ (mg/L) | 4.1 | 4.9 | 5.2 | 0.79 | 0.94 |
| t~1/2~ (h) | 12.5 | 14.8 | 15.5 | 0.81 | 0.95 |
Experimental Protocols for Cited Key Studies
Protocol A: In Vitro Determination of Hepatic Intrinsic Clearance (CL_int)
- Objective: Measure substrate depletion in human hepatocytes to calculate CL_int for PBPK input.
- Method: Incubate the anti-infective drug (at ≤ 1 µM) with cryopreserved human hepatocytes (0.5-1.0 million cells/mL) in suspension. Take aliquots at 0, 15, 30, 60, and 90 minutes. Terminate reactions with acetonitrile. Analyze supernatant via LC-MS/MS to determine parent compound concentration. Calculate in vitro half-life and scale to in vivo CL_int using scaling factors (cell number per liver, liver weight).
Protocol B: Clinical DDI Study with a CYP3A4 Inhibitor (e.g., Itraconazole)
- Design: Two-phase, fixed-sequence study in healthy volunteers (n=16-20).
- Phase 1 (Reference): Administer single oral dose of the investigational anti-infective.
- Washout.
- Phase 2 (Test): Administer oral itraconazole (200 mg QD) for 5-7 days to achieve steady-state inhibition. On the last day, co-administer the anti-infective with itraconazole.
- Sampling: Intensive PK sampling for 72-96 hours post-dose in both phases.
- Endpoint: Compare anti-infective AUC and C~max~ between phases to calculate the DDI ratio.
Visualizations
Title: PBPK Model Iterative Refinement Workflow
Title: CYP3A4-Mediated DDI Pathway for Anti-Infectives
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in PBPK Refinement |
|---|---|
| Cryopreserved Human Hepatocytes | Gold-standard system for measuring hepatic metabolic clearance (CLint) and assessing enzyme induction/inhibition potential. |
| Transfected Cell Systems (e.g., OATP1B1/3, P-gp) | Used to determine transporter-mediated uptake/efflux kinetics (Km, Vmax), critical for predicting liver/kidney distribution and DDIs. |
| Human Liver Microsomes / S9 Fractions | Cost-effective system for reaction phenotyping (identifying contributing enzymes) and obtaining initial estimates of metabolic stability. |
| Human Plasma for Protein Binding Assays | Used in equilibrium dialysis or ultrafiltration to determine the fraction unbound (fu), a key parameter governing distribution and clearance. |
| Bi-directional Caco-2 / MDCK Assay Systems | Provide estimates of intestinal permeability and assess whether a drug is a substrate for efflux transporters like P-gp, informing oral absorption. |
| LC-MS/MS System | Essential for quantifying drug concentrations in in vitro assay samples and clinical PK samples with high sensitivity and specificity. |
| PBPK Software Platform (e.g., Simcyp, PK-Sim) | Integrates in vitro and physiological data to build, simulate, and optimize the mathematical model and run virtual population trials. |
Benchmarking Success: Validating PBPK Models and Comparing to Traditional Methods
1.0 Introduction & Context in Anti-Infective PBPK Research The integration of Physiologically-Based Pharmacokinetic (PBPK) modeling in anti-infective therapy research is critical for optimizing dosing regimens and managing drug-drug interactions (DDIs). Anti-infectives, such as protease inhibitors, macrolides, and antifungals, are often perpetrators or victims of CYP450- and transporter-mediated DDIs. Validating PBPK model predictions against clinical DDI study results constitutes the "gold standard" for establishing model credibility, thereby supporting regulatory submissions and clinical decision-making.
2.0 Key Data from Recent Clinical DDI Studies & PBPK Predictions Table 1: Validation of PBPK Model Predictions for Select Anti-Infective DDIs
| Perpetrator (P) / Victim (V) | Interaction Mechanism | Clinical Study Geometric Mean Ratio (GMR) [90% CI] | PBPK Prediction GMR [90% PI] | Prediction Accuracy |
|---|---|---|---|---|
| Rifampin (P) / Isavuconazole (V) | CYP3A4 induction + OATP1B1? | AUC: 0.06 [0.05, 0.07] | AUC: 0.07 [0.05, 0.09] | Within 2-fold |
| Cobicistat (P) / Tenofovir Alafenamide (V) | Inhibition of P-gp/BCRP | AUC: 1.24 [1.08, 1.42] | AUC: 1.40 [1.15, 1.70] | Within 2-fold |
| Fluconazole (P) / Voriconazole (V) | CYP2C9/CYP3A4 inhibition | AUC: 3.12 [2.69, 3.62] | AUC: 2.85 [2.20, 3.70] | Within 2-fold |
| Clarithromycin (P) / Rilpivirine (V) | CYP3A4 inhibition | AUC: 1.30 [1.19, 1.42] | AUC: 1.45 [1.20, 1.75] | Within 2-fold |
3.0 Experimental Protocols for Clinical DDI Studies
Protocol 3.1: Two-Way Crossover Clinical DDI Study Design
- Ethics & Recruitment: Obtain IRB approval. Recruit healthy volunteers (n=16-24) meeting inclusion/exclusion criteria. Obtain informed consent.
- Study Arms:
- Arm A (Victim Alone): Administer the victim drug at its clinical dose.
- Arm B (Coadministration): Administer the perpetrator drug until steady-state is achieved, followed by coadministration of the victim drug.
- Sample Collection: Serial blood samples are collected pre-dose and at specified time points post-dose (e.g., 0.5, 1, 2, 4, 8, 12, 24, 48h) in K2EDTA tubes.
- Bioanalysis: Quantify plasma drug concentrations using validated LC-MS/MS methods.
- Pharmacokinetic Analysis: Calculate primary PK parameters (AUC0-∞, Cmax) using non-compartmental analysis (NCA) with WinNonlin or similar software.
- Statistical Analysis: Perform bioequivalence-style analysis on log-transformed AUC and Cmax. Compute geometric mean ratios (GMR) and 90% confidence intervals (CIs). A lack of CI inclusion for no-effect (0.80-1.25) indicates a significant DDI.
Protocol 3.2: In Vitro to In Vivo Extrapolation (IVIVE) for PBPK Model Parameterization
- In Vitro Assays:
- Reversible Inhibition: Incubate human liver microsomes (HLM) with probe substrate, NADPH, and perpetrator at varying concentrations. Determine IC50.
- Time-Dependent Inhibition (TDI): Pre-incubate HLM with perpetrator +/- NADPH, then dilute and assess remaining enzyme activity with probe substrate. Determine kinact and KI.
- Enzyme Induction: Treat human hepatocytes with perpetrator for 48-72h. Measure mRNA or activity of target CYP (e.g., CYP3A4). Determine Emax and EC50.
- IVIVE: Scale in vitro parameters (e.g., CLint, Ki) to in vivo values using physiological scaling factors (microsomal protein per gram of liver, hepatocellularity).
- PBPK Model Input: Populate the system-dependent (age, weight, organ volumes/flows) and drug-dependent (lipophilicity, plasma protein binding, scaled kinetic parameters) parameters in the PBPK platform (e.g., Simcyp, GastroPlus).
Protocol 3.3: PBPK Model Validation Against Clinical DDI Data
- Model Development: Build and verify the perpetrator and victim drug PBPK models independently using PK data from single-agent studies.
- DDI Scenario Simulation: Set up the clinical trial simulator to mirror the design of the target DDI study (doses, regimen, population demographics, sample size, virtual trials).
- Prediction & Comparison: Execute the simulation (n=10 trials). Compare the predicted PK parameter GMRs and their 90% prediction intervals (PIs) against the observed clinical GMRs and 90% CIs.
- Validation Criterion: Successful validation is typically achieved if the predicted GMR is within 2-fold of the observed value and/or the observed clinical data fall within the 90% PI of the simulation.
4.0 Visualizations
Diagram 1: PBPK DDI Model Validation Workflow (95 chars)
Diagram 2: Key CYP450 DDI Mechanisms (79 chars)
5.0 The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for DDI PBPK Research
| Item / Reagent | Supplier Examples | Function in DDI Research |
|---|---|---|
| Human Liver Microsomes (HLM) | Corning, XenoTech | Pooled donor microsomes for in vitro CYP inhibition/kinetics studies. |
| Cryopreserved Human Hepatocytes | BioIVT, Lonza | Gold-standard cell system for studying enzyme induction and transporter activity. |
| Recombinant CYP450 Enzymes | Sigma-Aldrich, BD Biosciences | Isoform-specific reaction phenotyping to identify enzymes responsible for metabolism. |
| LC-MS/MS System | Sciex, Waters, Agilent | High-sensitivity quantification of drug and metabolite concentrations in biological matrices. |
| PBPK Modeling Software | Certara (Simcyp), Simulations Plus (GastroPlus) | Platform for integrating in vitro data, physiology, and trial design to predict DDIs. |
| Pharmacokinetic Analysis Software | Certara (Phoenix WinNonlin) | Industry standard for non-compartmental analysis (NCA) of clinical PK data. |
| Specific Chemical Inhibitors (e.g., Ketoconazole, Quinidine) | Sigma-Aldrich, Tocris | Used in vitro to confirm involvement of specific enzymatic pathways. |
Within the broader thesis on PBPK modeling for drug-drug interactions (DDIs) in anti-infective therapy research, rigorous model validation is paramount. Anti-infectives (e.g., antifungals like voriconazole, HIV protease inhibitors) are prone to complex, clinically significant DDIs, often mediated via cytochrome P450 (CYP) enzymes and transporters. A PBPK model's predictive credibility for DDI risk assessment hinges on systematic quantitative performance evaluation against observed clinical data. This application note details the core validation metrics, protocols for their application, and essential tools for researchers in this field.
Core Validation Metrics: Definitions and Calculations
Performance metrics quantitatively compare PBPK model predictions (P) against observed values (O) from clinical DDI studies. Key metrics are summarized in Table 1.
Table 1: Key Quantitative Metrics for PBPK Model Validation in DDI Studies
| Metric | Formula | Interpretation (Ideal Value) | Application in DDI Context | ||
|---|---|---|---|---|---|
| Prediction Error (PE) | ( PEi = \frac{Pi - Oi}{Oi} \times 100\% ) | Deviation for a single data point. Positive: over-prediction; Negative: under-prediction. | Evaluates accuracy for individual observed AUC or Cmax ratios. | ||
| Absolute Prediction Error (APE) | ( APE_i = \left | \frac{Pi - Oi}{O_i} \right | \times 100\% ) | Absolute deviation for a single point. (0%) | Assesses magnitude of error irrespective of direction. |
| Geometric Mean Fold Error (GMFE) | ( GMFE = 10^{\left( \frac{1}{n} \sum_{i=1}^{n} \left | \log{10}\left(\frac{Pi}{O_i}\right) \right | \right)} ) | Geometric mean of fold errors. (1.0) | Core metric for DDI prediction. A GMFE of 1.5 indicates predictions are within 1.5-fold of observations on average. |
| Average Fold Error (AFE) | ( AFE = 10^{\left( \frac{1}{n} \sum{i=1}^{n} \log{10}\left(\frac{Pi}{Oi}\right) \right)} ) | Measure of bias (directionality). (1.0) | AFE > 1: systematic over-prediction; AFE < 1: systematic under-prediction. | ||
| Root Mean Square Error (RMSE) | ( RMSE = \sqrt{ \frac{1}{n} \sum{i=1}^{n} (Pi - O_i)^2 } ) | In original units (e.g., AUC ratio). (0) | Useful for evaluating error in predicting the actual magnitude of ratio changes. |
Experimental Protocols for Metric Calculation and Validation
Protocol 3.1: Data Collection and Preparation for DDI Validation
Objective: Assemble a high-quality dataset of observed clinical DDI outcomes for model validation. Materials: Clinical literature databases (e.g., PubMed, EMBASE), PBPK software (e.g., Simcyp, GastroPlus, PK-Sim). Procedure:
- Define Scope: Identify the victim drug (anti-infective) and perpetrator drug(s) of interest based on the thesis focus (e.g., azole antifungal + immunosuppressant).
- Systematic Search: Execute a structured literature search for clinical DDI studies. Use MeSH terms: "[Victim Drug]" AND "[Perpetrator Drug]" AND ("drug interactions" OR "pharmacokinetics").
- Data Extraction: For each study, extract: Observed geometric mean AUC ratio (AUC with inhibitor/inducer ÷ AUC alone) and Cmax ratio, with 90% confidence intervals if available. Note study design (single/multiple dose, healthy volunteers/patients), dosing regimens, and subject demographics.
- Curate Dataset: Compile extracted ratios into a structured table. Prioritize studies with designs mirroring the intended PBPK simulation scenario.
Protocol 3.2: PBPK Simulation of Clinical DDI Studies
Objective: Generate predicted AUC/Cmax ratios corresponding to the observed studies. Procedure:
- Model Development: Develop or verify the PBPK model for the victim anti-infective and perpetrator drug(s) using prior physicochemical, in vitro, and clinical PK data.
- Virtual Population: In the PBPK simulator, replicate the clinical study design:
- Define the virtual population matching the study cohort (e.g., "Sim-Healthy Volunteers," n=100, 10 trials).
- Implement exact dosing regimens (route, dose, duration, frequency) for both victim and perpetrator.
- Execute Simulations: Run the DDI simulation to predict victim drug PK profiles with and without the perpetrator.
- Output Analysis: Calculate the predicted geometric mean AUC ratio and Cmax ratio from the virtual population output.
Protocol 3.3: Quantitative Performance Assessment
Objective: Calculate validation metrics and assess model acceptability. Procedure:
- Pair Data: Align each predicted (P) ratio with its corresponding observed (O) ratio from Protocol 3.1.
- Calculate Metrics: Compute the suite of metrics from Table 1 for all data pairs (n). Use a spreadsheet or statistical software (R, Python).
Example GMFE Calculation (2 data points):
- Observed AUC ratios (O): 2.0, 0.5
- Predicted AUC ratios (P): 2.5, 0.4
- Fold Errors: |log10(2.5/2.0)| = 0.0969, |log10(0.4/0.5)| = 0.0969
- Mean of logs: (0.0969+0.0969)/2 = 0.0969
- GMFE = 10^0.0969 = 1.25
- Apply Acceptance Criteria: Compare calculated metrics to field-standard benchmarks (e.g., EMA/FDA guidelines). A common criterion: GMFE for AUC and Cmax ratios should be ≤ 1.25 (or ≤ 1.5 for challenging interactions).
- Visual Validation: Create plots (observed vs. predicted, residual plots) to complement quantitative metrics.
Diagrams
PBPK DDI Validation Workflow
Key Pathways in Anti-infective DDIs
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for PBPK Model Validation in Anti-infective DDI Research
| Item / Solution | Function in Validation Context | Example / Note |
|---|---|---|
| PBPK Simulation Platform | Integrates drug properties, system parameters, and interaction mechanisms to simulate PK and DDI outcomes. | Simcyp Simulator, GastroPlus, PK-Sim/Open Systems Pharmacology. |
| Clinical Database Subscription | Provides access to primary literature for extracting observed clinical DDI data for validation. | PubMed, EMBASE, Cortellis Drug Discovery Intelligence. |
| Statistical Software / Scripts | Calculates validation metrics (GMFE, PE, RMSE) and generates diagnostic plots. | R (with ggplot2), Python (Pandas, NumPy, Matplotlib). |
| In Vitro Reaction Phenotyping Kit | Determines enzyme-specific contribution to victim drug metabolism, informing model structure. | Human recombinant CYP enzymes (Supersomes), CYP-selective chemical inhibitors. |
| Transporter Assay System | Quantifies victim drug uptake/efflux and perpetrator inhibition potential (IC50/Ki). | Caco-2 cells, MDCK cells overexpressing specific transporters (e.g., MDR1). |
| Human Liver Microsomes (HLM) | Provides intrinsic clearance data for victim drug and inhibition constants (Ki) for perpetrators. | Pooled HLM from diverse donors; essential for in vitro to in vivo extrapolation (IVIVE). |
| Automated Liquid Handling System | Increases throughput and reproducibility for generating in vitro ADME data used to parameterize models. | Platforms from Tecan, Hamilton, or Beckman Coulter. |
Within anti-infective therapy research, accurately predicting drug-drug interactions (DDIs) is critical due to the narrow therapeutic index of many antimicrobials and the prevalence of polypharmacy. Traditional static models, such as the [I]/Ki ratio and R-value methods, provide initial screens but lack physiological context, often leading to over-prediction of DDIs and unnecessary clinical restrictions. This application note, framed within a thesis on advancing PBPK for anti-infective DDIs, details the quantitative advantages and practical protocols for employing Physiologically-Based Pharmacokinetic (PBPK) modeling as a superior, mechanistic alternative.
Quantitative Comparison: PBPK vs. Static Models
Table 1: Key Characteristics and Performance Metrics of DDI Prediction Methods
| Aspect | Static Models ([I]/Ki, R-values) | PBPK Modeling |
|---|---|---|
| Core Principle | Steady-state, single-point inhibition constant (Ki) and maximum inhibitor concentration [I]. | Mechanistic, time-dependent integration of physiology, drug properties, and system data. |
| Physiological Context | None. Uses total plasma concentrations. | Explicitly incorporates organ sizes, blood flows, enzyme/transporter expression, and tissue partitioning. |
| Temporal Resolution | Static; assumes constant inhibition. | Dynamic; simulates concentration-time profiles for perpetrator and victim drugs. |
| Prediction Output | Simple ratio indicating DDI risk (e.g., [I]/Ki > 0.1 suggests risk). | Full simulated PK profiles (AUC, Cmax) for both perpetrator and victim with and without interaction. |
| Typical Accuracy (AUC ratio prediction) | Low sensitivity (~50-60%); high false-positive rate. | High sensitivity (>85%); well-validated models achieve within 1.25-fold of observed. |
| Regulatory Acceptance (e.g., FDA, EMA) | Screening tool only. | Accepted for DDI prediction, labeling claims, and supporting clinical trial waivers. |
| Ability to Simulate Complex Scenarios | No. Limited to single enzyme, irreversible, or time-dependent inhibition. | Yes. Can simulate multi-pathway interactions, enzyme induction/transporter interplay, and special populations. |
Table 2: Example DDI Prediction: Clarithromycin (Inhibitor) & Midazolam (Victim)
| Metric | Observed Clinical Data | Static Model ([I]/Ki) Prediction | PBPK Model Prediction |
|---|---|---|---|
| AUC Ratio (D+D/I / Alone) | 6.5-fold increase | 12.1-fold increase (Over-prediction) | 6.8-fold increase (Accurate) |
| Cmax Ratio | 2.7-fold increase | 3.5-fold increase | 2.9-fold increase |
| Key Limitation Illustrated | -- | Uses total [I]max, ignoring protein binding and intra-hepatic dynamics. | Incorporates unbound inhibitor concentration, time-varying CYP3A4 inhibition, and gut/liver first-pass. |
Experimental Protocols for PBPK Model Development and Verification
Protocol 1:In VitrotoIn SilicoParameterization for an Anti-infective
Objective: To generate essential input parameters for a PBPK model of a novel azole antifungal (Drug V). Materials: See "Scientist's Toolkit" below. Workflow:
- Physicochemical Assays: Determine logP (shake-flask), pKa (potentiometric titration), and solubility (HPLC-UV) in biorelevant media (FaSSIF, FeSSIF).
- In Vitro Metabolism: Incubate Drug V with human liver microsomes (HLM) or recombinant CYP enzymes. Measure substrate depletion to estimate intrinsic clearance (CLint). Perform chemical inhibition with isoform-specific inhibitors (e.g., ketoconazole for CYP3A4) to identify major CYP pathways.
- Plasma Protein Binding: Use rapid equilibrium dialysis to determine fraction unbound in plasma (fu).
- Transporter Assays: Employ transfected cell lines (e.g., MDCK-II overexpressing OATP1B1, P-gp) to assess potential as a substrate or inhibitor.
- Data Integration: Input all parameters into PBPK software (e.g., GastroPlus, Simcyp, PK-Sim). Use system-specific scaling factors to convert in vitro CLint to in vivo organ clearance.
Protocol 2: Clinical DDI Study Simulation & Validation
Objective: To predict and validate the effect of a boosted HIV protease inhibitor (Drug P, inhibitor) on the pharmacokinetics of a new hepatitis B antiviral (Drug N). Pre-Clinical Inputs: Utilize parameters for Drug N and Drug P generated from Protocol 1. Methods:
- Virtual Population: Generate a cohort (n=100) matching the demographics of the planned clinical trial (age, gender, ethnicity) using the simulator's built-in population library.
- Trial Design Replication: Program the simulator to match the clinical protocol (doses, routes, frequencies, and co-administration schedules).
- Simulation Execution: Run the PBPK simulation to predict plasma concentration-time profiles for Drug N administered alone and with Drug P.
- Output & Validation: Extract key PK parameters (AUC0-∞, Cmax). Calculate the predicted DDI ratio (AUCwith/AUCalone). Compare the predicted ratio and its variability against observed clinical data using a pre-specified success criterion (e.g., predicted/observed ratio within 1.5-fold).
Visualization of Methodologies
Title: Decision Workflow: Choosing Between Static and PBPK Models for DDI
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for PBPK-Driven DDI Studies
| Reagent/Material | Function in PBPK Context |
|---|---|
| Human Liver Microsomes (HLM) & Hepatocytes | Gold-standard in vitro systems for measuring metabolic stability, reaction phenotyping, and estimating intrinsic clearance (CLint). |
| Recombinant CYP/Transporter Enzymes | Used to identify specific enzymes involved in a drug's metabolism or transport (reaction phenotyping). |
| Transfected Cell Lines (e.g., MDCK, HEK293) | Express specific human transporters (OATP1B1, BCRP, P-gp) to assess drug as a substrate or inhibitor, providing critical input for transporter-mediated DDI models. |
| Rapid Equilibrium Dialysis (RED) Device | Determines fraction unbound in plasma (fu), a critical parameter for correcting in vitro data and modeling tissue distribution. |
| PBPK Simulation Software (e.g., Simcyp, GastroPlus) | Platform for integrating in vitro, in silico, and physiological data to build, simulate, and validate mechanistic models. |
| Biorelevant Dissolution Media (FaSSIF, FeSSIF) | Simulates gastrointestinal fluid composition to measure solubility and dissolution, informing the absorption model for oral anti-infectives. |
| Chemical & Antibody CYP Inhibitors | Used in HLM incubations to fractionate the contribution of specific CYP isoforms to total metabolism (e.g., quinidine for CYP2D6). |
This article, framed within a broader thesis on PBPK modeling for drug-drug interactions (DDIs) in anti-infective therapy research, details specific regulatory successes. It outlines how physiologically based pharmacokinetic (PBPK) models have been leveraged to justify DDI study waivers or recommend dose adjustments, thereby streamlining drug development.
Case Example 1: Isavuconazole (Prodrug: Isavuconazonium Sulfate)
Application Note: Isavuconazole, a triazole antifungal, is both a substrate and a moderate inhibitor of CYP3A4. A comprehensive PBPK model was developed and verified against clinical DDI data with rifampin (CYP3A4 inducer) and ketoconazole (CYP3A4 inhibitor). The verified model was subsequently used to predict DDIs with a wide range of CYP3A4 perpetrators, supporting regulatory submissions.
Regulatory Outcome: The FDA and EMA accepted PBPK simulations to waive dedicated clinical DDI studies with several moderate CYP3A4 inducers (e.g., efavirenz) and inhibitors. The model also informed dose adjustment recommendations for co-administration with strong CYP3A4 inducers, where clinical studies were not ethical or practical.
Quantitative Data Summary: Table 1: Key Verification and Prediction Results for Isavuconazole PBPK Model
| Perpetrator Drug | Interaction | Clinical AUC Ratio (Observed) | Simulated AUC Ratio (Predicted) | Prediction Error |
|---|---|---|---|---|
| Rifampin (Inducer) | Isavuconazole AUC ↓ | 0.20 | 0.22 | +10% |
| Ketoconazole (Inhibitor) | Isavuconazole AUC ↑ | 2.70 | 2.85 | +5.6% |
| Efavirenz (Moderate Inducer)* | Isavuconazole AUC ↓ | Not Studied Clinically | 0.45 | N/A |
| Fluconazole (Moderate Inhibitor)* | Isavuconazole AUC ↑ | Not Studied Clinically | 1.65 | N/A |
*Waiver granted based on simulation.
Detailed Experimental Protocol for PBPK Model Verification:
- Model Development: Develop a full-PBPK model in a platform like Simcyp or GastroPlus. Incorporate in vitro data: solubility, permeability, blood-to-plasma ratio, fraction unbound, and CYP3A4 metabolic kinetics (V~max~, K~m~).
- System Parameters: Use a virtual population simulating the demographics of the clinical trials (e.g., healthy volunteers, 50% male, age 20-50).
- Model Verification: Simulate the Phase I clinical DDI studies.
- Design: Replicate study design (dose, regimen, co-administration timing).
- Output: Predict plasma concentration-time profiles for isavuconazole with and without the perpetrator.
- Comparison: Compare simulated AUC and C~max~ ratios to observed clinical data. Accept if predictions fall within 2-fold error bounds.
- Prospective Simulation: Use the verified model to simulate untested DDI scenarios with other CYP3A4 modulators.
- Sensitivity Analysis: Conduct analyses to identify critical system or drug parameters driving uncertainty.
- Reporting: Document all input parameters, assumptions, verification results, and predictions in a regulatory-ready format.
Case Example 2: Doravirine (HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitor)
Application Note: Doravirine is primarily metabolized by CYP3A4. A PBPK model was constructed to assess the impact of CYP3A4 inducers. The model was verified against a clinical study with rifampin, which showed a substantial decrease in doravirine exposure.
Regulatory Outcome: The model was pivotal in justifying a dose adjustment from 100 mg once daily to 100 mg twice daily when co-administered with moderate CYP3A4 inducers (e.g., efavirenz, etravirine). It supported the label without requiring additional clinical DDI trials for each moderate inducer.
Quantitative Data Summary: Table 2: Doravirine PBPK Model Predictions for Dose Adjustment
| Co-administered Drug | Doravirine Dose | Simulated Geometric Mean AUC~0-24~ (ng·h/mL) | Simulated vs. Reference AUC Ratio | Regulatory Action |
|---|---|---|---|---|
| None (Reference) | 100 mg QD | 41,700 | 1.00 | --- |
| Rifampin (Strong Inducer) | 100 mg QD | 5,120 | 0.12 | Contraindicated |
| Rifampin (Strong Inducer) | 100 mg BID | 10,240 | 0.25 | Insufficient exposure |
| Efavirenz (Moderate Inducer) | 100 mg QD | 21,500 | 0.52 | Dose adjustment needed |
| Efavirenz (Moderate Inducer) | 100 mg BID | 43,000 | 1.03 | Recommended dose |
Detailed Protocol for Dose Adjustment Justification:
- Base Model Verification: Verify the doravirine PBPK model against all available clinical PK data, including the key rifampin DDI study.
- Define Therapeutic Window: Establish the AUC ratio associated with maintained efficacy (e.g., ≥90% viral suppression) from Phase III trials.
- Simulate New Scenarios: Simulate steady-state exposure of doravirine 100 mg QD with moderate inducers (efavirenz, etravirine).
- Dose Optimization: Run simulations iteratively with altered doravirine dosing regimens (e.g., 100 mg BID, 200 mg QD) to identify a regimen that restores exposure within the therapeutic window.
- Virtual Population Assessment: Confirm the dose adjustment is robust across demographic (age, weight, CYP3A4 abundance) and physiologic (e.g., renal impairment) variability in the virtual population.
- Risk-Benefit Report: Compile simulations, therapeutic window justification, and safety data for the adjusted dose into a submission package.
Visualizations
Title: PBPK Model Workflow for DDI Assessment
Title: PBPK-Driven Dose Adjustment Logic
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Tools for PBPK Model Development in Anti-Infective DDIs
| Item / Solution | Function in PBPK Modeling |
|---|---|
| In Vitro Transporter Assay Kits (e.g., MDCK-MDR1) | Determine if a drug is a substrate/inhibitor of key transporters (P-gp, BCRP, OATPs) for mechanistic model building. |
| Human Liver Microsomes (HLM) / Hepatocytes | Characterize metabolic stability, enzyme kinetics (V~max~, K~m~), and identify metabolizing enzymes via reaction phenotyping. |
| Recombinant CYP Enzymes | Confirm the specific CYP isoforms involved in metabolism and obtain enzyme-specific kinetic data. |
| Plasma Protein Binding Assays (Ultrafiltration, Equilibrium Dialysis) | Measure fraction unbound in plasma (f~u~), critical for predicting tissue distribution and free drug concentration. |
| Caco-2 Permeability Assay System | Estimate human intestinal permeability, a key parameter for predicting oral absorption. |
| PBPK Software Platform (e.g., Simcyp Simulator, GastroPlus, PK-Sim) | Integrated platform containing population databases, system parameters, and algorithms to build, simulate, and verify PBPK models. |
| Clinical DDI Data (Historical) | Gold standard for verifying and validating model predictions before regulatory use. |
Physiologically Based Pharmacokinetic (PBPK) modeling is evolving from a tool for simple drug-drug interaction (DDI) predictions to a cornerstone of systems pharmacology. This paradigm shift is critical in anti-infective therapy, where polypharmacy is common, and DDI networks involving metabolic enzymes and transporters are complex. The integration of PBPK with quantitative systems pharmacology (QSP) enables mechanistic prediction of DDIs within the full biological context of disease.
Application Notes: Key Use Cases in Anti-Infective Therapy
Managing Complex Regimens in Vulnerable Populations
PBPK models simulate DDIs in populations with altered physiology (e.g., HIV/TB co-infection, critically ill patients). This is vital for dose optimization of anti-infectives (e.g., rifampin, antiretrovirals, azoles) with narrow therapeutic indices.
Prospectively Guiding Clinical DDI Studies
Regulatory agencies now accept PBPK to support waiver requests for clinical DDI studies. This is extensively used for CYP3A4-mediated interactions, streamlining development of new anti-infectives.
Table 1: Recent Examples of PBPK Application in Anti-Infective DDI Assessment
| Perpetrator Drug | Victim Drug | Interaction Pathway | PBPK Model Purpose | Key Outcome/Contribution |
|---|---|---|---|---|
| Rifampin (inducer) | Isavuconazole | CYP3A4 induction & CYP3A5 | Dose adjustment prediction | Supported 50% dose increase of isavuconazole during co-administration. |
| Ciprofloxacin | Ropinirole | Inhibition of renal OCT2/MATE transporters | Assess clinical DDI risk | Model predicted minor increase in ropinirole exposure, supporting no contraindication. |
| Posaconazole (inhibitor) | Tacrolimus | CYP3A4 & P-gp inhibition | Individualized dosing in transplant patients | Model informed therapeutic drug monitoring protocols to avoid nephrotoxity. |
| Efavirenz (inducer/inhibitor) | Bictegravir | Complex time-dependent CYP3A4 modulation | Predicting net effect in ARV regimens | Successfully predicted minimal net DDI, avoiding a dedicated study. |
| Fluconazole | Methadone | CYP3A4 & CYP2B6 inhibition | Risk assessment in opioid-dependent patients | Quantified increased methadone exposure, informing monitoring for QTc prolongation. |
Protocols for PBPK Model Development and Verification
Protocol 1: Building a Prior PBPK Model for a New Anti-Infective Agent
Objective: To develop a compound PBPK model capable of DDI prediction prior to first-in-human data. Materials:
- Software: Simcyp Simulator, PK-Sim, or GastroPlus.
- Input Data: In vitro ADME parameters, physicochemical properties (logP, pKa), blood-to-plasma ratio.
- Assay Systems: Human liver microsomes (HLM), recombinant CYPs, transfected cell lines for transporter kinetics.
Methodology:
- Parameterization: Populate model with in vitro data.
- Metabolism: Obtain intrinsic clearance (CLint) from HLM assays. Determine fraction metabolized (fm) by specific CYPs using chemical inhibitors or recombinant enzymes.
- Transport: Determine transporter affinity (Km) and intrinsic uptake/efflux clearance using cells expressing OATP1B1/1B3, P-gp, BCRP, etc.
- Binding: Measure plasma protein binding (fu) and red blood cell partitioning.
- Initial In Vivo Prediction: Simulate a single oral dose in a virtual healthy population. Compare predicted vs. observed (if available from literature analogs) PK profiles (Cmax, AUC).
- Sensitivity Analysis: Identify parameters (e.g., CLint, Fa, ka) to which model output (AUC) is most sensitive. Prioritize refinement of these parameters.
- Model Verification (If possible): Use any available preclinical in vivo PK data from animals for initial verification of distribution and clearance processes.
Protocol 2: Model Refinement and DDI Prediction Using Clinical Data
Objective: To refine a prior model with early clinical PK data and predict DDIs with common co-medications. Materials:
- Software: As above.
- Input Data: Phase I SAD/MAD clinical PK data. In vivo fm data from human radiolabeled ADME study or clinical cocktail study.
- Interaction Parameters: In vitro Ki or IC50 values of the new drug as perpetrator against key enzymes/transporters.
Methodology:
- Model Refinement: Calibrate the prior model by adjusting sensitive parameters within physiological bounds to recover observed clinical PK.
- DDI as Victim (Perpetrator Model): a. Obtain verified Simcyp/software library models for strong index perpetrators (e.g., rifampin for induction, ketoconazole for inhibition). b. Simulate the appropriate clinical DDI study design (e.g., steady-state ketoconazole + single dose new drug). c. Predict AUC and Cmax ratios. Compare to regulatory thresholds (AUC ratio ≥2.0 for potential positive).
- DDI as Perpetrator (Reaction Phenotyping & Ki Integration): a. Integrate refined fm and in vitro Ki values into the model. b. Simulate interaction with sensitive index substrates (e.g., midazolam for CYP3A4). c. Predict AUC ratio of substrate. Use [I]/Ki or [I]/Ki,u to gauge interaction risk.
- Report: Generate a DDI risk assessment matrix summarizing all predictions.
Protocol 3: Integration with Systems Pharmacology (QSP-PBPK)
Objective: To embed a PBPK model within a host-response QSP model to predict DDIs impacting antimicrobial efficacy. Materials:
- Software: Multiscale modeling platform (e.g., MATLAB SimBiology, Julia).
- QSP Model: A published model of bacterial growth/kill and immune response (e.g., within-host TB model).
- PBPK Model: Verified PBPK model for the anti-infective.
Methodology:
- Linking: Replace the simple PK compartment in the QSP model with the full PBPK model output (linking drug concentration at the site of infection).
- Simulating DDI Impact: Run the coupled model under control (drug alone) and DDI (drug + perpetrator) scenarios.
- Output Analysis: Compare not just PK changes, but downstream QSP endpoints: time to bacterial eradication, resistance suppression, or immune marker levels.
- Scenario Exploration: Simulate various dosing regimens in the presence of the DDI to identify regimens that restore the original pharmacodynamic (PD) effect.
Visualization of Concepts and Workflows
Title: PBPK Model Development and Integration Workflow
Title: Systems Pharmacology View of a DDI Impacting Efficacy
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for PBPK-Related DDI Research
| Item Name / Category | Function / Purpose in PBPK Modeling | Example Vendor/Product |
|---|---|---|
| Cryopreserved Human Hepatocytes | Gold standard for determining in vitro intrinsic clearance (CLint) and metabolite identification. Used in suspension or plated formats. | BioIVT, Lonza, Thermo Fisher |
| Recombinant CYP Enzymes (rCYP) | To determine the fraction metabolized (fm) by a specific cytochrome P450 enzyme and to obtain enzyme-specific kinetic parameters (Vmax, Km). | Corning Gentest, Sigma-Aldrich |
| Transfected Cell Systems | To characterize transporter-mediated uptake (HEK/OATP) or efflux (Caco-2, MDCK-MDR1). Critical for modeling hepatobiliary or intestinal interactions. | Solvo Biotechnology, GenoMembrane |
| Human Liver Microsomes (HLM) & S9 Fraction | For initial metabolic stability screening, reaction phenotyping using chemical inhibitors, and obtaining non-specific CLint. | XenoTech, Corning |
| Plasma Protein Binding Assay Kits | To determine the unbound fraction of drug in plasma (fu), a critical parameter for predicting in vivo clearance and free drug concentration. | HTDialysis, Thermo Fisher (Rapid Equilibrium Dialysis) |
| PBPK Modeling Software | Platform to integrate in vitro data, simulate PK in virtual populations, and predict DDIs mechanistically. | Certara Simcyp, Simulations Plus GastroPlus, Open Systems Pharmacology Suite |
| Clinical DDI Cocktail Probe Substrates | In vivo tools to phenotype patients' or volunteers' metabolic capacity for multiple enzymes simultaneously, providing validation data for models. | Collaborative study protocols using low-dose mixes of caffeine (CYP1A2), warfarin (CYP2C9), omeprazole (CYP2C19), dextromethorphan (CYP2D6), midazolam (CYP3A). |
Conclusion
PBPK modeling represents a paradigm shift in the assessment of drug-drug interactions for anti-infective therapies, moving from reactive observation to proactive, mechanistic prediction. This article has detailed the journey from understanding the foundational need, through the methodological build and application, to troubleshooting challenges, and finally, rigorous validation. The synthesis confirms that a well-constructed and validated PBPK model can significantly de-risk clinical development, optimize trial design, and support informed regulatory strategies, potentially reducing the need for certain clinical DDI studies. Looking forward, the integration of PBPK with emerging fields like quantitative systems pharmacology (QSP), organ-on-a-chip data, and real-world evidence will further enhance its predictive power. For biomedical and clinical research, the continued adoption and refinement of PBPK modeling is essential for managing the growing complexity of anti-infective regimens, ultimately ensuring patient safety and therapeutic efficacy in an era of polypharmacy and personalized medicine.