Bridging the Gap: A Comprehensive Guide to In Vitro-In Vivo Correlation of Anti-Infective Efficacy
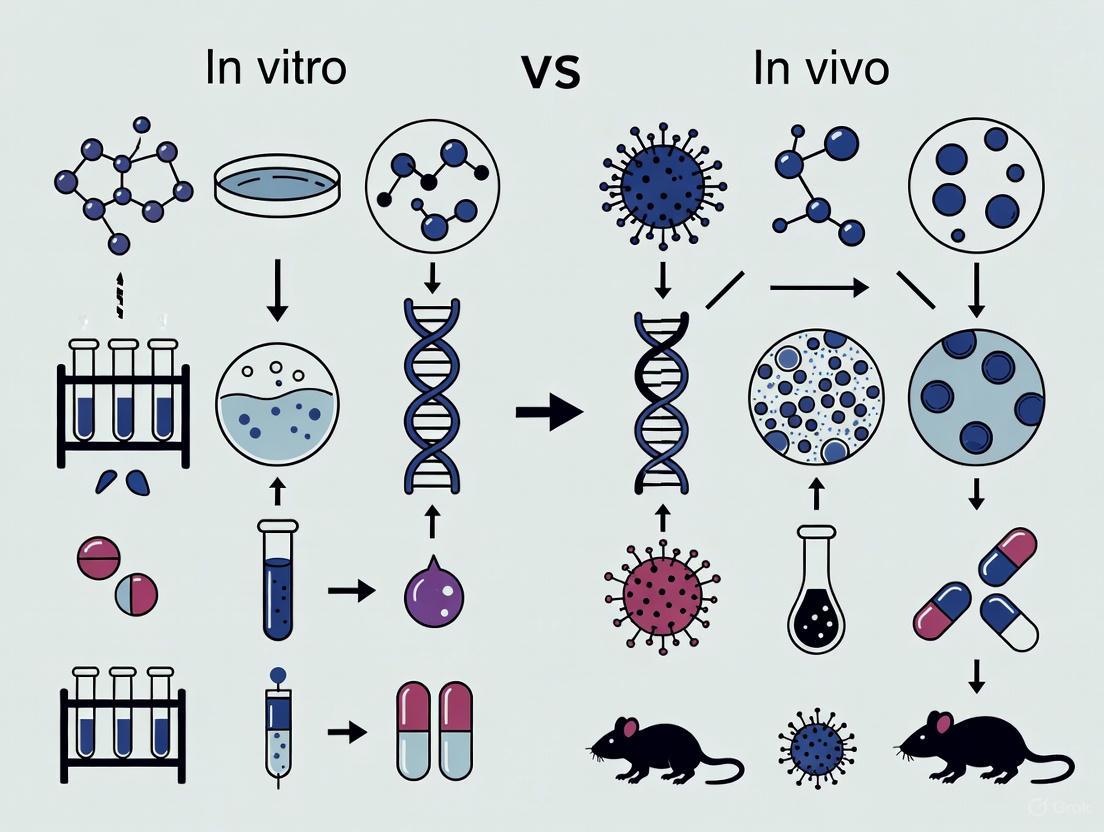
Establishing a reliable correlation between in vitro and in vivo efficacy (IVIVC) is a critical yet challenging endeavor in anti-infective drug development.
Bridging the Gap: A Comprehensive Guide to In Vitro-In Vivo Correlation of Anti-Infective Efficacy
Abstract
Establishing a reliable correlation between in vitro and in vivo efficacy (IVIVC) is a critical yet challenging endeavor in anti-infective drug development. This article provides a comprehensive overview for researchers and drug development professionals, exploring the foundational principles of IVIVC and the significant obstacles posed by physiological complexity and biofilm-related infections. It delves into advanced methodological approaches, including sophisticated in vitro models and PK/PD modeling, which are enhancing predictive power. The content further addresses troubleshooting common discrepancies and offers strategies for model optimization. Finally, it examines validation frameworks and comparative analyses of successful IVIVC case studies across different anti-infective classes, synthesizing key takeaways to guide future research and improve the translation of preclinical findings to clinical success.
The Core Challenge: Why Predicting In Vivo Efficacy from In Vitro Data is So Difficult
In the relentless pursuit of novel anti-infective therapies, researchers navigate a critical transition between controlled laboratory studies and the complex reality of living systems. The chasm separating in vitro (in an artificial environment) and in vivo (within a living organism) results is a pivotal focus in antimicrobial development, where promising laboratory findings often fail to translate into clinical efficacy. This guide objectively compares the performance and outcomes of anti-infective agents across these two environments, framing the analysis within the broader context of in vitro-in vivo correlation (IVIVC). Understanding these fundamental differences is not merely an academic exercise but a practical necessity for designing more predictive experiments, accelerating drug development, and ultimately delivering effective treatments to patients grappling with antimicrobial-resistant infections.
Fundamental Environmental Differences
The disparity between in vitro and in vivo results stems from profound differences in environmental complexity. The in vitro environment is a simplified, controlled system designed to isolate specific biological interactions. In contrast, the in vivo environment is an interconnected network of biological systems that introduces numerous variables absent in laboratory settings.
The diagram above illustrates the fundamental environmental divide. In vitro systems lack the dynamic pharmacokinetic/pharmacodynamic (PK/PD) profiles present in living organisms, where drugs experience absorption, distribution, metabolism, and excretion [1] [2]. The absence of a functional immune system in vitro eliminates potential synergistic antimicrobial effects, as even potent peptides like Ctn[15-34] must function without immune assistance [2]. Furthermore, in vitro models typically employ single-species cultures that ignore polymicrobial interactions and biofilm communities commonly encountered in clinical infections [3]. The simplified growth media used in laboratories cannot replicate the complex composition of biological fluids, which contain proteins that bind drugs, enzymes that degrade therapeutics, and variable pH levels that alter antimicrobial activity [4] [2].
Quantitative Comparison of Anti-infective Efficacy
The environmental differences between laboratory and living systems manifest as quantifiable disparities in antimicrobial efficacy. The table below summarizes comparative data from recent studies demonstrating these gaps for various anti-infective agents.
Table 1: Comparative Efficacy of Anti-infective Agents: In Vitro vs. In Vivo Results
| Anti-infective Agent | Pathogen | In Vitro Efficacy (MIC) | In Vivo Efficacy | Key Disparity Factors |
|---|---|---|---|---|
| Cefiderocol [1] | Pseudomonas aeruginosa | Susceptible (MIC ≤2 µg/mL) | 1-log kill at 24h; 2-log kill at 48h (murine thigh) | PK/PD parameters, immune component absence |
| Ceftolozane/Tazobactam [1] | Pseudomonas aeruginosa | Susceptible (MIC ≤2/4 µg/mL) | 1-log kill in 3/5 isolates at 24h (murine thigh) | Inoculum size, host-pathogen dynamics |
| Ctn[15-34] peptide [2] | Acinetobacter baumannii | Potent activity (low MIC) | Reduced bacterial load with gender-specific effects (murine) | Proteolytic stability, toxicity profiles |
| Ctn retroenantio analog [2] | Acinetobacter baumannii | Improved stability & activity in vitro | Toxic at 5-30 mg/kg; no efficacy (murine) | Unpredicted toxicity, biological recognition |
| Melia azedarach CuO NPs + Cefepime [5] | Multidrug-resistant Klebsiella pneumoniae | MIC: 1.92 µg/mL (synergistic) | 82% inhibition; improved histopathology (in vivo) | Immune modulation, tissue penetration |
| SK1260 Antimicrobial Peptide [6] | E. coli, S. aureus, K. pneumoniae, P. aeruginosa | MIC: 3.13-12.5 µg/mL | Reduced bacterial burden in organs; improved survival (murine) | Serum binding, biodistribution, immune effects |
The data reveals several critical patterns. First, efficacy magnitude disparities are common, as seen with cefiderocol which demonstrated more rapid killing profiles in vivo compared to ceftolozane/tazobactam despite similar in vitro susceptibility [1]. Second, unpredicted toxicity emerges in vivo for compounds showing excellent in vitro safety profiles, exemplified by the retroenantio analogs of Ctn peptides that proved toxic in mouse models despite promising laboratory data [2]. Third, the influence of host systems significantly modulates outcomes, as demonstrated by CuO nanoparticles that showed enhanced wound healing and immune regulation in vivo beyond their direct antimicrobial effects observed in vitro [5].
Experimental Protocols and Methodologies
Understanding the methodological approaches for evaluating anti-infectives in both environments is crucial for interpreting correlation data. This section details standard protocols used in recent studies.
In Vitro Susceptibility Testing Protocols
Minimum Inhibitory Concentration (MIC) Assay
- Procedure: Serial dilutions of antimicrobial agents in 96-well microtiter plates containing appropriate culture medium are inoculated with approximately 10^5 CFU/mL of bacterial suspension [6]. Positive (bacterial growth) and negative (sterile) controls are included. After incubation at 37°C for 16-24 hours, the MIC is determined as the lowest concentration showing no visible growth [6].
- Key Reagents: Cation-adjusted Mueller-Hinton broth, log-phase bacterial cultures, sterile physiological saline for dilutions.
- Data Analysis: MIC values are reported in µg/mL, with experiments typically performed in triplicate for statistical reliability.
Time-Kill Kinetics Assay
- Procedure: Bacterial suspensions are exposed to antimicrobial concentrations (typically 0.5×, 1×, and 5× MIC) and incubated at 37°C [6]. Aliquots are collected at predetermined time intervals (0, 1, 2, 3, 4, 5, 12, and 24 hours), serially diluted, and plated on nutrient agar. After 24 hours of incubation, colony-forming units (CFU) are enumerated.
- Data Analysis: Results are expressed as log10 CFU/mL versus time, with bactericidal activity defined as ≥3-log reduction from initial inoculum.
Biphasic Dissolution System for IVIVC
- Procedure: This advanced system contains aqueous (buffer, 300 mL) and organic (octanol, 200 mL) phases saturated through stirring at 37°C [7]. Drug formulations are introduced into the aqueous phase, with samples simultaneously collected from both phases at multiple time points. The system evaluates both dissolution and partitioning kinetics, providing a more biorelevant assessment for poorly soluble compounds [7].
In Vivo Efficacy Assessment Protocols
Murine Thigh Infection Model
- Procedure: Mice are rendered neutropenic via cyclophosphamide administration (150 mg/kg, 4 days and 1 day before infection) [1]. Thighs are inoculated with approximately 10^6 CFU of bacteria. Human-simulated regimens of antimicrobials are administered starting 2 hours post-infection. Thighs are harvested at predetermined times (24, 48, 72 hours), homogenized, and plated for bacterial quantification.
- Key Parameters: Change in bacterial density from baseline (log10 CFU/thigh), with translational endpoints of 1- and 2-log10 kill [1].
Systemic Infection and Survival Models
- Procedure: Mice are infected intraperitoneally with lethal inocula of pathogens, frequently supplemented with mucin to enhance infection establishment [2]. Test articles are administered prophylactically or therapeutically at various doses. Animals are monitored for morbidity, mortality, and clinical scores for several days. For bacterial burden studies, target organs (liver, spleen, kidney, lung) are harvested, homogenized, and plated for bacterial quantification [6].
The experimental workflow demonstrates the sequential approach to anti-infective evaluation, where promising in vitro candidates progress to increasingly complex in vivo models, culminating in IVIVC analysis that bridges the two environments.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful navigation of the in vitro-in vivo continuum requires specialized reagents and materials tailored to anti-infective research. The table below catalogues critical solutions and their applications.
Table 2: Essential Research Reagent Solutions for Anti-infective Efficacy Studies
| Reagent/Material | Application | Function in Research |
|---|---|---|
| Cation-Adjusted Mueller-Hinton Broth [6] | In vitro susceptibility testing | Standardized medium for MIC and time-kill assays ensuring reproducible results |
| Biphasic Dissolution System [7] | IVIVC for poorly soluble drugs | Simultaneously evaluates drug dissolution and partitioning kinetics using aqueous and organic phases |
| OptiPrep Density Gradient Medium [3] | OMV isolation and purification | Separates bacterial outer membrane vesicles from other cellular components for mechanistic studies |
| XTT Cell Proliferation Kit II [4] | Metabolic activity assessment | Measures bacterial viability and metabolic activity after antimicrobial exposure through colorimetric detection |
| Propidium Iodide Stain [6] | Membrane integrity testing | Evaluates membrane disruption by antimicrobial peptides through fluorescence detection of DNA binding |
| Hank's Balanced Salt Solution (HBSS) [4] | pH measurement studies | Maintains physiological ion balance while measuring alkalinity of intracanal medicaments over time |
| Porcine Mucin [2] | In vivo infection models | Enhances bacterial virulence in animal models by providing protective matrix and immunosuppression |
| Lipolysis Assay Components [8] | Lipid formulation evaluation | Simulates intestinal digestion to predict in vivo performance of lipid-based drug formulations |
Implications for Anti-infective Drug Development
The discordance between in vitro and in vivo environments presents both challenges and opportunities for anti-infective development. The failure of compounds like retroenantio AMP analogs despite promising in vitro profiles underscores the perils of over-relying on simplified systems [2]. Conversely, the successful translation of SK1260 peptide, which demonstrated correlative in vitro and in vivo efficacy against multiple pathogens, highlights the value of robust preclinical models [6].
The emergence of advanced technologies offers promising avenues for bridging the gap. Biorelevant dissolution systems incorporating lipid digestion processes improve predictions for lipid-based formulations [8]. Bacterial outer membrane vesicle research provides insights into resistance mechanisms and potential therapeutic applications [3]. Furthermore, the integration of artificial intelligence and machine learning approaches enables more sophisticated analysis of complex datasets, potentially identifying patterns that predict in vivo performance from in vitro data [9].
Ultimately, recognizing the fundamental differences between these environments enables researchers to design more predictive experiments, interpret results more critically, and make better decisions about which candidates merit progression to clinical trials. This understanding is paramount in an era of escalating antimicrobial resistance, where efficient translation of laboratory discoveries to effective patient therapies is an urgent global priority.
Bacterial biofilms are structured communities of cells encased in a self-produced extracellular matrix and represent one of the most widespread forms of microbial life [10]. In clinical settings, biofilm-associated infections are responsible for approximately 80% of all microbial infections [10]. These include persistent conditions such as endocarditis, osteomyelitis, infections related to cystic fibrosis, and those occurring on medical implants [10]. A hallmark of biofilm-related infections is their recalcitrance to antimicrobial treatment, which frequently leads to chronic infections, implant failure, and increased mortality [10]. This resistance profile observed in clinical settings often starkly contrasts with the susceptibility patterns determined through standard in vitro antimicrobial susceptibility testing (AST), creating a significant hurdle in anti-infective drug development [11] [12]. This guide examines the mechanisms behind these discrepancies and compares conventional and emerging models for evaluating anti-biofilm efficacy.
Comparative Analysis: Planktonic vs. Biofilm Antimicrobial Susceptibility
Traditional AST methods, such as broth microdilution, primarily assess the susceptibility of free-floating (planktonic) bacteria by determining parameters like the Minimal Inhibitory Concentration (MIC), which is the lowest concentration that prevents visible growth [13]. However, these methods fail to accurately predict the efficacy of antimicrobials against biofilm-associated infections. The table below summarizes the key differences in how antimicrobials act on these two distinct bacterial lifestyles.
Table 1: Key Differences Between Planktonic and Biofilm Antimicrobial Susceptibility
| Feature | Planktonic Cells (Standard AST) | Biofilm Cells |
|---|---|---|
| Primary Metric | Minimal Inhibitory Concentration (MIC) [13] | Minimal Duration for Killing (MDK), e.g., MDK99 [13] |
| Defining Phenotype | Resistance (ability to grow at concentrations above the MIC) [13] | Tolerance (ability to survive exposure to concentrations above the MIC without regrowing) [13] |
| Underlying Mechanisms | Target modification, enzymatic inactivation, efflux pumps [13] | Reduced penetration, metabolic heterogeneity, persister cells, matrix protection [13] [14] |
| Response to Treatment | Typically eradicated by concentrations at or above the MIC | Often survive transient treatment, leading to relapse [13] |
Core Mechanisms of Biofilm-Mediated Treatment Failure
The reduced susceptibility of biofilms is not attributable to a single mechanism but rather a multi-faceted barrier. The following sections detail the primary contributing factors, which often act in concert.
Physical and Chemical Barriers
The Extracellular Polymeric Substance (EPS) matrix acts as a primary barrier by hindering the penetration of antimicrobial agents into the deeper layers of the biofilm [13] [14]. For example, the negatively charged polysaccharide alginate in Pseudomonas aeruginosa biofilms can bind and retain the aminoglycoside antibiotic tobramycin [14]. Similarly, an increase in extracellular DNA (eDNA) concentration in Staphylococcus epidermidis biofilms reduces the penetration of vancomycin [14]. Furthermore, the matrix can accumulate antibiotic-degrading enzymes, such as β-lactamase, effectively inactivating the drug before it reaches its target [14].
Physiological Heterogeneity
Gradients of oxygen, nutrients, and waste products within the biofilm create a spectrum of microenvironments [13]. This leads to a heterogeneous population of cells with vastly different metabolic states. A key consequence is the presence of slowly growing or dormant cells [13] [12]. Since many antibiotics are only effective against actively growing bacteria, these dormant subpopulations exhibit profound tolerance and can repopulate the biofilm once antibiotic pressure is removed [13].
Evolutionary Dynamics and Adaptive Responses
The spatially structured nature of biofilms, with its gradients of antimicrobial agents, creates 'sanctuaries' where drug concentrations are sub-lethal [13]. These sanctuaries can act as 'stepping stones,' allowing bacterial populations to acquire resistance mutations sequentially, a process that would be impossible in a homogeneous, high-concentration environment [13]. Biofilms also exhibit increased mutation rates compared to planktonic cultures, partly due to oxidative stress, which accelerates genetic adaptation [13]. Additionally, the high cell density and presence of eDNA in the matrix facilitate Horizontal Gene Transfer (HGT), promoting the spread of resistance genes [13].
The following diagram illustrates the core mechanisms contributing to biofilm antimicrobial tolerance.
Advanced Experimental Models for Biofilm Research
To bridge the gap between in vitro predictions and in vivo outcomes, researchers are developing more sophisticated models that better mimic the in vivo environment.
In Vitro Flow Models for Anaerobic Co-Culture
Studying host-microbe interactions in the gut has been challenging due to the conflicting oxygen requirements of human cells and obligate anaerobic microbiota. A recent 2025 model addresses this by using a dual-flow channel system with an integrated anaerobization unit [15]. This system maintains stable oxygen levels below 1% in the apical (luminal) channel while supplying oxygen to the intestinal cells from the basolateral side, enabling long-term co-culture of human epithelium with obligate anaerobes like Clostridioides difficile [15]. This model has demonstrated the persistence of C. difficile following vancomycin treatment, replicating a key clinical challenge [15].
Experimental Evolution in Biofilms
Experimental evolution, where bacterial populations are repeatedly exposed to antimicrobial treatment in controlled laboratory settings, provides powerful insights into resistance development. When performed in biofilms, these studies reveal that spatial structure significantly influences evolutionary trajectories [13]. Population fragmentation within the biofilm leads to independently evolving subpopulations, fostering greater genetic diversity and allowing for the fixation of beneficial mutations that might be lost in a well-mixed planktonic culture [13].
Parameters for Predicting Resistance Emergence
Beyond the MIC, several in vitro parameters can help forecast a compound's potential to select for resistance, which is crucial at the hit-to-lead stage of drug development [16].
Table 2: Key In Vitro Parameters for Forecasting Resistance Development
| Parameter | Definition | Utility in Prediction |
|---|---|---|
| Mutant Prevention Concentration (MPC) | The antibiotic concentration that prevents the growth of the least susceptible, single-step mutant in a large bacterial population [16]. | Helps define the upper limit of the mutant selection window (MSW); dosing above MPC may suppress resistance. |
| Mutant Selection Window (MSW) | The concentration range between the MIC of the wild-type strain and the MPC [13]. | Antibiotic concentrations within this window enrich for resistant mutants. |
| Frequency of Spontaneous Mutant Selection (FSMS) | The ratio of resistant colony-forming units (CFUs) to the total number of CFUs plated on antibiotic-containing media [16]. | Quantifies the probability that a single-step resistant mutant will arise spontaneously. |
| Minimal Selective Concentration (MSC) | The lowest antibiotic concentration at which the growth rate of a resistant mutant equals that of the wild-type strain [16]. | Defines the lower boundary of the selective window, including at sub-MIC levels. |
The relationship between these parameters and selective pressure is visualized below.
The Scientist's Toolkit: Essential Reagents and Models
Selecting appropriate experimental tools is critical for generating clinically relevant data on anti-biofilm efficacy.
Table 3: Key Research Reagent Solutions for Biofilm Studies
| Tool Category | Specific Examples | Function & Application |
|---|---|---|
| Biofilm Quantification | Crystal Violet (CV) Staining, Resazurin Viability Staining, Colony Forming Unit (CFU) Enumeration [14] | CV measures total biomass; Resazurin assesses metabolic activity; CFU counts culturable cells. Each measures a different aspect of biofilms. |
| Advanced In Vitro Models | Dual-flow Channel Systems, Organ-on-a-Chip, Anaerobization Units [15] | Create physiologically relevant environments with controlled oxygen gradients and fluid shear stress for host-microbe co-culture. |
| Matrix Targeting Agents | DNase (degrades eDNA), Dispersin B (degrades polysaccharide), N-Acetylcysteine (breaks disulfide bonds) [10] [17] | Enzymatically degrade specific components of the EPS matrix to disrupt biofilm structure and enhance antimicrobial penetration. |
| Anti-Virulence Agents | Quorum Sensing Inhibitors (e.g., AHL analogs), Anti-adhesion coatings [10] [17] | Target bacterial cell-to-cell communication and surface attachment without exerting direct lethal pressure, potentially reducing resistance selection. |
The disconnect between in vitro activity and in vivo efficacy of antimicrobial agents against biofilms remains a significant obstacle in anti-infective development [11]. This discrepancy is rooted in the fundamental physiological, structural, and evolutionary differences between planktonic and biofilm communities. Relying solely on traditional AST, which is designed for planktonic bacteria, leads to poor predictive value for biofilm-associated infections. Success in this field requires the adoption of more sophisticated, physiologically relevant in vitro models that incorporate flow, host cells, and controlled microenvironments, alongside a focus on anti-biofilm specific parameters like the MPC and MDK. By integrating these advanced tools and concepts into the drug development pipeline, researchers can better forecast clinical outcomes and design more effective strategies to combat persistent biofilm infections.
The efficacy of anti-infective therapies is traditionally predicted using in vitro susceptibility tests, such as broth microdilution. However, these routine methods often fail to predict clinical outcomes for device-related infections (DRIs), as they do not account for the biofilm phenotype of bacteria. This guide compares the performance of standard planktonic susceptibility testing with advanced biofilm susceptibility methods, framing the analysis within the critical thesis of in vitro versus in vivo correlation.
Comparison of Susceptibility Testing Methodologies
The following table compares the core methodologies, their underlying principles, and key performance metrics.
Table 1: Methodological Comparison of Planktonic vs. Biofilm Susceptibility Testing
| Feature | Routine Planktonic Testing (e.g., Broth Microdilution) | Advanced Biofilm Susceptibility Testing (e.g., Calgary Biofilm Device) |
|---|---|---|
| Bacterial Phenotype | Free-floating (Planktonic) | Surface-attached community (Biofilm) |
| Key Output Metric | Minimum Inhibitory Concentration (MIC) | Minimum Biofilm Eradication Concentration (MBEC) |
| Correlation with DRI Outcomes | Poor. Routinely underestimates the antibiotic concentration required for eradication. | Strong. Better predicts the need for higher doses or combination therapies. |
| Experimental Data (S. aureus vs. Oxacillin) | MIC: 0.5 µg/mL (Susceptible) | MBEC: >256 µg/mL (Resistant) |
| Underlying Reason for Discrepancy | Tests bacteria in a vulnerable, non-adherent state. | Accounts for matrix protection, reduced metabolic activity, and persister cells. |
Experimental Protocols
Protocol 1: Standard Broth Microdilution for MIC Determination This protocol is the reference method for determining the Minimum Inhibitory Concentration (MIC) against planktonic bacteria.
- Preparation: Prepare a logarithmic dilution series (e.g., two-fold) of the antibiotic in a suitable broth medium (e.g., Mueller-Hinton Broth) in a 96-well microtiter plate.
- Inoculation: Standardize a bacterial suspension to approximately 5 x 10^5 CFU/mL in the same broth. Add a equal volume of this suspension to each well of the antibiotic dilution series. Include growth control (bacteria, no antibiotic) and sterility control (broth only) wells.
- Incubation: Incubate the plate under optimal conditions for the test organism (e.g., 35±2°C for 16-20 hours).
- Result Interpretation: The MIC is the lowest concentration of antibiotic that completely inhibits visible growth of the organism.
Protocol 2: Calgary Biofilm Device (CBD) for MBEC Determination This protocol is used to determine the Minimum Biofilm Eradication Concentration (MBEC), which measures the concentration required to kill biofilm-encased bacteria.
- Biofilm Formation: Inoculate a specialized CBD lid (with 96 pegs) into a microtiter plate containing a standardized bacterial suspension. Incubate the assembly under static conditions for a defined period (e.g., 24-48 hours) to allow biofilms to form on the pegs.
- Biofilm Maturation: After incubation, gently rinse the peg lid in a neutral buffer to remove non-adherent planktonic cells.
- Antibiotic Challenge: Transfer the peg lid to a new "challenge" plate containing a two-fold dilution series of the antibiotic in broth. Incubate for a further 24 hours.
- Biofilm Disruption and Viability Assessment: Remove the peg lid, rinse again to remove residual antibiotic, and then transfer it to a "recovery" plate containing a neutral buffer. Sonicate or vortex the plate to dislodge and disaggregate biofilm cells from the pegs. Serially dilute the recovered suspension and spot-plate onto agar plates to enumerate viable colony-forming units (CFUs).
- Result Interpretation: The MBEC is the lowest concentration of antibiotic that results in a ≥3-log10 reduction (99.9% kill) in viable biofilm bacteria compared to the growth control.
Visualizing the In Vitro / In Vivo Disconnect
The following diagrams illustrate the logical workflow of the testing methods and the core biological reasons for the failure of routine tests.
Workflow: Routine MIC Test
Biofilm Resistance Mechanisms
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Biofilm Susceptibility Research
| Item | Function |
|---|---|
| Calgary Biofilm Device (CBD) | A standardized peg-lid apparatus for high-throughput cultivation and testing of biofilms. |
| Crystal Violet Stain | A simple dye used for the semi-quantitative assessment of total biofilm biomass. |
| Resazurin Viability Stain | An oxidation-reduction indicator used to measure metabolic activity within biofilms. |
| Mueller-Hinton Broth | The standardized growth medium specified for antimicrobial susceptibility testing. |
| Tryptic Soy Agar (TSA) | A general-purpose growth medium used for the enumeration of viable bacteria (CFU counting). |
| Polystyrene Microplates | The standard platform for broth microdilution (planktonic) and biofilm assays. |
| Cation-Adjusted Mueller-Hinton Broth (CAMHB) | A refined broth that ensures consistent cation concentrations, critical for reliable antibiotic activity, particularly with aminoglycosides. |
In anti-infective drug development, a profound disconnect often exists between promising in vitro results and clinical efficacy. This translational gap stems primarily from the failure of simplistic laboratory models to account for the intricate physiological complexity of living organisms. While in vitro susceptibility testing provides essential initial data on antimicrobial activity, it occurs in an environment largely devoid of host immunity, pharmacokinetic (PK) variables, and biological barriers that determine drug distribution to infection sites. The transition from static in vitro conditions to dynamic in vivo systems introduces multifaceted challenges including protein binding, tissue penetration limitations, variable metabolic conditions, and active host immune responses that collectively modulate therapeutic outcomes. Understanding these complex interactions is critical for accurate prediction of clinical efficacy and optimization of dosing regimens for anti-infective agents.
Key Physiological Barriers in Anti-Infective Efficacy
The Critical Role of Host Immunity
In vivo infection models demonstrate that host immune status dramatically influences antibacterial efficacy, a factor completely absent in standard in vitro testing. The neutropenic murine thigh infection model, a cornerstone of anti-infective pharmacodynamics, explicitly controls for this variable by rendering mice immunocompromised before infection [18]. This model allows researchers to isolate drug effects from immune-mediated clearance, providing a standardized platform for comparing antimicrobial activity under defined conditions. However, this represents only one point on the spectrum of immune competence that clinicians encounter in human populations.
The immune system interacts with anti-infective therapies through multiple mechanisms:
- Synergistic clearance: Immune cells such as neutrophils and macrophages may work in concert with antibiotics to eliminate pathogens more effectively than either component alone.
- Altered pharmacokinetics: Inflammation can change blood flow, capillary permeability, and protein binding, thereby modifying drug distribution to infection sites.
- Differential expression in immunocompromised states: The efficacy of bacteriostatic versus bactericidal agents may vary significantly depending on host immune status, with bactericidal agents generally preferred in immunocompromised patients.
Pharmacokinetic and Pharmacodynamic Considerations
Pharmacokinetics (what the body does to the drug) and pharmacodynamics (what the drug does to the body) collectively determine anti-infective efficacy in vivo. The integration of these disciplines through PK/PD modeling has become essential for translating in vitro activity to in vivo effectiveness [18]. Key principles include:
- Time-dependent versus concentration-dependent killing: Different antibiotic classes exhibit distinct killing patterns that inform optimal dosing strategies. Beta-lactams typically show time-dependent activity, requiring concentrations to remain above the MIC for extended periods, while aminoglycosides display concentration-dependent killing, benefiting from higher peak concentrations.
- Post-antibiotic effects: Some antibiotics continue suppressing bacterial growth even after concentrations fall below the MIC, an phenomenon only observable in dynamic systems.
- Protein binding impact: Only unbound drug molecules can exert antimicrobial activity, making protein binding a critical determinant of efficacy that varies between in vitro media and in vivo conditions [19].
Tissue Distribution and Penetration Barriers
Perhaps the most significant translational challenge lies in achieving adequate drug concentrations at the site of infection, which often differs substantially from plasma levels measured in pharmacokinetic studies [20]. Multiple factors complicate tissue distribution:
- Blood-tissue barriers: Capillary physiology varies significantly across tissues, with permeability coefficients differing by orders of magnitude between organs [20]. The blood-brain barrier represents the most extreme example, but other tissues like bone, prostate, and abscess cavities also present significant penetration challenges.
- Interstitial fluid as the target site: For most bacterial infections, the relevant compartment is the interstitial space fluid (ISF) where extracellular pathogens reside [20]. Drug concentrations in this compartment often differ markedly from plasma levels due to capillary wall resistance, pH partitioning, and active transport mechanisms.
- Methodological misconceptions: Traditional approaches to measuring tissue concentrations through homogenization provide misleading data by admixing intracellular, extracellular, and vascular compartments [20]. More sophisticated techniques like microdialysis now enable direct measurement of unbound drug concentrations in the ISF, providing more pharmacologically relevant data [19].
Table 1: Key Physiological Factors Creating Discrepancies Between In Vitro and In Vivo Anti-infective Efficacy
| Physiological Factor | In Vitro Simplification | In Vivo Complexity | Impact on Efficacy |
|---|---|---|---|
| Host Immunity | Absent | Variable immune competence | Can synergize with or compensate for drug activity |
| Protein Binding | Often standardized or ignored | Variable binding to plasma and tissue proteins | Reduces free, active drug concentrations |
| Tissue Penetration | Uniform drug distribution | Barriers based on capillary structure and physiology | Creates concentration gradients between plasma and infection sites |
| Pathophysiology | Optimal, uniform growth conditions | Altered pH, oxygen tension, nutrient availability | Affects bacterial growth rate and antimicrobial susceptibility |
Comparative Analysis: Cefiderocol versus Ceftolozane/Tazobactam
Experimental Methodology and Translational Model
A recent comparative study exemplifies the importance of physiological considerations when evaluating anti-infective efficacy [1]. This investigation employed a 72-hour murine thigh infection model against five clinical difficult-to-treat Pseudomonas aeruginosa isolates to compare cefiderocol and ceftolozane/tazobactam. The methodology incorporated several key physiological elements:
- Human-simulated regimens: The study utilized human-simulating regimens of ceftolozane/tazobactam (2/1 g IV q8h) and cefiderocol (2 g IV q8h) to replicate human pharmacokinetic profiles in the murine model, enhancing translational relevance [1].
- Immunocompromised host: The neutropenic murine model controlled for variability in immune response, allowing isolation of drug-specific effects [1].
- Extended duration: The 72-hour timeframe enabled assessment of both initial killing and potential resistance development over multiple drug exposures.
- Bacterial density measurements: Efficacy was quantified as change in bacterial density from starting inoculum, with comparison to translational endpoints of 1- and 2-log₁₀ kill [1].
Efficacy and Killing Kinetics Comparison
Despite both agents demonstrating susceptibility against the tested isolates in vitro, significant differences emerged in their in vivo performance profiles [1]:
- Rate of killing: Cefiderocol achieved 1-log₁₀ kill against all five isolates by 24 hours, while ceftolozane/tazobactam reached this endpoint in only three of five isolates.
- Magnitude of effect: Cefiderocol produced 2-log₁₀ kill in all isolates by 48 hours, whereas ceftolozane/tazobactam required 72 hours to achieve this level of killing in four isolates.
- Bacterial eradication: The cefiderocol treatment group showed 17% bacterial eradication versus 8% in the ceftolozane/tazobactam group after exposure to human-simulated regimens.
Table 2: In Vivo Efficacy Comparison Between Cefiderocol and Ceftolozane/Tazobactam Against Difficult-to-Treat P. aeruginosa in a Murine Thigh Infection Model [1]
| Efficacy Parameter | Cefiderocol | Ceftolozane/Tazobactam |
|---|---|---|
| 1-log₁₀ Kill at 24h | 5/5 isolates | 3/5 isolates |
| 2-log₁₀ Kill at 48h | 5/5 isolates | 0/5 isolates |
| 2-log₁₀ Kill at 72h | 5/5 isolates | 4/5 isolates |
| Bacterial Eradication | 17% of cultures | 8% of cultures |
| Resistance Development | Not detected | Not detected |
Mechanistic Insights and Physiological Advantages
The superior performance of cefiderocol in this model can be attributed to its unique mechanism of action that specifically addresses physiological challenges:
- Siderophore functionality: Cefiderocol utilizes the bacterial iron transport system to actively cross outer membranes, bypassing traditional porin channels and efflux pumps that often mediate resistance in Pseudomonas aeruginosa [1].
- Enhanced tissue penetration: The siderophore mechanism may facilitate improved penetration through physiological barriers that limit distribution of other beta-lactams.
- Stability against degradation: Cefiderocol demonstrates greater stability against beta-lactamases, including extended-spectrum beta-lactamases (ESBLs) and carbapenemases, which are frequently expressed by difficult-to-treat pathogens.
Methodological Framework for Translation
Integrated PK/PD Modeling Approaches
Model-based translation represents a sophisticated alternative to traditional PK/PD index approaches that better accounts for physiological complexity [18]. The development of a mechanism-based PK/PD model for the FabI inhibitor afabicin illustrates this paradigm:
- In vitro model development: A PK/PD model was built using 162 static in vitro time-kill curves evaluating afabicin desphosphono against 21 Staphylococcus aureus strains [18].
- Bacterial dynamics modeling: The model incorporated two bacterial states (growing/susceptible and dormant/non-susceptible) to account for heterogeneous subpopulations with different susceptibility profiles [18].
- Translational prediction: When combined with a mouse PK model, parameters estimated from in vitro data successfully predicted in vivo bacterial counts at 24 hours within ±1 log margin for most dosing groups without parameter re-estimation [18].
- Parameter refinement: Subsequent estimation from in vivo data revealed that the EC₅₀ was 38-45% lower in vivo compared to in vitro, highlighting important physiological differences in drug activity [18].
Measuring Therapeutically Relevant Concentrations
Accurate assessment of drug exposure at infection sites requires methodological sophistication beyond traditional plasma monitoring:
- Unbound drug concentrations: Only unbound drug molecules can exert antimicrobial activity, making protein binding a critical determinant of efficacy [19]. Techniques for direct measurement of free drug concentrations include microdialysis, ultrafiltration, and equilibrium dialysis.
- Interstitial fluid sampling: For most extracellular infections, the relevant compartment is the interstitial space fluid where pathogens reside [20]. Microdialysis enables direct measurement of unbound antibiotic concentrations in this compartment through semi-permeable membranes implanted in tissues [19].
- Accounting for protein binding variability: Protein binding exhibits concentration-dependent behavior for some antibiotics (e.g., ceftriaxone, ertapenem) and may change in disease states due to alterations in plasma protein concentrations [19].
Table 3: Essential Research Reagent Solutions for Evaluating Physiological Complexity in Anti-infective Studies
| Research Tool | Function | Application Context |
|---|---|---|
| Neutropenic Murine Thigh Model | Standardized assessment of in vivo efficacy independent of host immunity | Preclinical PK/PD studies for antibacterial agents [18] |
| Microdialysis Systems | Direct measurement of unbound drug concentrations in interstitial fluid | Tissue penetration studies for antibiotics with poor distribution [19] |
| HepG2 Cell Line | In vitro model for mRNA vaccine translation and protein expression | Potency assessment for mRNA-based vaccines [21] |
| Mechanism-Based PK/PD Models | Mathematical frameworks describing time course of antibiotic effects | Translation from in vitro time-kill data to in vivo efficacy prediction [18] |
Implications for Anti-Infective Drug Development
Optimizing Dosing Strategies
Understanding physiological complexity enables more rational design of dosing regimens that maximize efficacy while minimizing toxicity and resistance development:
- Tissue penetration-informed dosing: For antibiotics with poor penetration to specific sites of infection (e.g., central nervous system, prostate), higher doses or alternative administration routes may be necessary to achieve therapeutic concentrations [20].
- Protein binding considerations: Highly protein-bound antibiotics may require dose adjustments in patients with altered protein binding capacity (e.g., malnutrition, liver disease, critical illness) [19].
- Immune status-adapted therapy: Immunocompromised patients often require more aggressive antibacterial therapy, including higher doses, combination therapy, or preferentially bactericidal agents.
Future Directions and Innovations
Several emerging approaches show promise for better incorporating physiological complexity into anti-infective development:
- Advanced infection models: More sophisticated in vitro and in vivo models that better mimic human physiology, including organ-on-a-chip systems and humanized mouse models.
- Imaging technologies: Non-invasive imaging techniques that enable real-time monitoring of infection progression and drug distribution in living organisms.
- Systems pharmacology: Integrated modeling approaches that incorporate host-pathogen-drug interactions across multiple biological scales from molecular to whole-organism levels.
- Biomarker development: Identification of biomarkers that predict tissue penetration and efficacy before clinical trials.
The translational gap between in vitro activity and in vivo efficacy remains a fundamental challenge in anti-infective development. The comparative analysis of cefiderocol and ceftolozane/tazobactam demonstrates how agents with similar in vitro susceptibility profiles can exhibit meaningfully different performance in physiologically relevant models. Successful translation requires meticulous attention to host immunity, pharmacokinetic variability, and tissue distribution barriers that collectively determine drug exposure at infection sites. Advanced modeling approaches that integrate in vitro and in vivo data, coupled with sophisticated sampling techniques that measure therapeutically relevant unbound drug concentrations at target sites, provide powerful tools for bridging this translational divide. As anti-infective development confronts the escalating challenge of antimicrobial resistance, accounting for physiological complexity will become increasingly critical for optimizing therapeutic outcomes and extending the utility of existing agents.
Advanced Models and PK/PD Strategies for Robust Correlation
The Trajectory of In Vitro Models: From Static Wells to Dynamic Microphysiological Systems
The journey of in vitro models began with simple two-dimensional (2D) cell cultures in static well plates. While these systems provided a foundational platform for basic research, they suffered from significant limitations, including distorted cell morphology, loss of tissue-specific functions, and an inability to replicate the complex three-dimensional (3D) architecture and dynamic cellular interactions found in living tissues [22]. This lack of physiological relevance often led to experimental data that poorly predicted human clinical responses, creating a critical gap in drug development [23].
The pressing need for more predictive models, coupled with ethical imperatives to reduce animal testing (as reinforced by policies like the FDA Modernization Act 2.0), has accelerated the development of advanced systems [22]. This evolution has progressed through several key stages:
- 2D Static Cultures: The traditional workhorse, useful for high-throughput screening but lacking physiological context.
- 3D Organoids and Spheroids: These models form cell aggregates that better mimic the spatial organization and some functional aspects of native tissues, bridging the gap between 2D cultures and whole organs [22].
- Organ-on-a-Chip (OoC) Systems: Representing the current state-of-the-art, OoC technology leverages microfluidics to create dynamic, perfused microenvironments. These chips can house miniature engineered tissues under conditions that mimic physiological fluid flow, mechanical forces (such as cyclic stretch in lungs and peristalsis in guts), and complex tissue-tissue interfaces [24] [23].
The integration of patient-derived organoids (PDOs) into OoC systems is a particularly powerful advancement. PDOs retain key genetic, phenotypic, and pathological features of the parent tumor, achieving high predictive accuracy—for example, >87% in colorectal cancer drug-response studies [22]. This convergence of biology and engineering has given rise to sophisticated microphysiological systems (MPS) that are transforming the study of human physiology, disease mechanisms, and drug efficacy.
Quantitative Comparison of In Vitro Model Capabilities
The table below summarizes the key characteristics of different in vitro models, highlighting the evolution in physiological relevance and application potential.
Table 1: Comparative Analysis of In Vitro Model Systems
| Feature | Traditional 2D Static Models | 3D Organoid/Spheroid Models | High-Throughput Organ-on-Chip (e.g., OrganoPlate) | Multi-Organ Chip Systems |
|---|---|---|---|---|
| Physiological Complexity | Low; monolayer culture, no 3D structure [22] | Moderate; 3D architecture, preserves some heterogeneity [22] | High; 3D tissue embedded in ECM, perfused tubules, apical/basolateral access [25] | Very High; multiple engineered tissues linked by vascular perfusion [23] |
| Throughput & Scalability | Very High (e.g., 384-well plates) | Moderate to High | High (40, 64, or 96 independent chips per plate) [25] | Low to Moderate; complex operation [25] |
| Dynamic Microenvironment | No; static culture | Limited; often static | Yes; continuous perfusion, controlled shear stress [25] | Yes; recirculating flow mimics systemic blood circulation [23] |
| Key Advantages | Simplicity, cost-effectiveness, high-throughput compatibility | Captures tumor heterogeneity, patient-specific [22] | Scalability for screening, direct compound access to tissue, no artificial membranes [25] | Studies inter-organ crosstalk, systemic drug PK/PD, and organism-level responses [23] |
| Primary Applications | Initial high-throughput drug screening, basic cell biology | Disease modeling, personalized therapy screening [22] | Complex tissue and disease modeling, transport and permeability assays, migration studies [25] | Preclinical assessment of drug safety, efficacy, and mechanistic toxicology [26] [23] |
Experimental Protocols for Key Assays in Anti-Infective Research
Establishing a correlation between in vitro potency and in vivo efficacy is a cornerstone of anti-infective development. The following protocols detail both traditional and advanced methods.
Protocol 1: Traditional Time-Kill Kinetics Assay
This method evaluates the temporal dynamics of antibacterial activity by tracking changes in bacterial concentration after antibiotic exposure [27].
Methodology:
- Inoculum Preparation: Prepare a bacterial suspension of approximately 10^5-10^6 CFU/mL in a suitable broth medium [27].
- Antibiotic Exposure: Add the antibiotic to the suspension at predetermined concentrations (e.g., 0.5x, 1x, 2x, and 4x the Minimum Inhibitory Concentration (MIC)).
- Incubation and Sampling: Incubate the culture under controlled conditions (e.g., 37°C). Take samples at regular intervals (e.g., 0, 2, 4, 6, and 24 hours).
- Viable Count Determination: Serially dilute each sample and plate it onto agar plates. After incubation, count the colony-forming units (CFU) to determine the number of viable bacteria at each time point.
- Data Analysis: Plot the log10 CFU/mL against time for each antibiotic concentration. The resulting time-kill curves show whether the antibiotic effect is bactericidal (≥3-log reduction in CFU/mL) or bacteriostatic [27].
Limitations: This assay is performed at a constant antibiotic concentration, which does not replicate the fluctuating concentrations seen in the human body. It also often lacks continuous nutrient supply and does not account for metabolites or host immune factors [27].
Protocol 2: Organ-on-a-Chip Model for Host-Pathogen Interaction and Drug Efficacy
This protocol leverages a perfused microfluidic system to create a human-relevant model for studying infections.
Methodology:
- Chip Priming and ECM Seeding:
- Use a microfluidic chip such as the OrganoPlate (3-lane 40 or 64) or Emulate Chip-S1 [25] [26].
- Inject an extracellular matrix (ECM) hydrogel, like collagen-I, into the central gel channel and allow it to polymerize.
- Flow culture medium through the two adjacent perfusion channels to condition the chip.
- Cell Seeding and Tissue Formation:
- Seed relevant epithelial or endothelial cells into the channels to form tissue barriers or perfused tubules. For instance, in a lung model, primary human airway epithelial cells can be cultured to create a mucociliary epithelium [23].
- Allow the tissues to differentiate and mature under perfusion for several days, often applying physiological cues like fluid shear stress or cyclic mechanical stretch.
- Infection and Drug Treatment:
- Introduce the pathogen (e.g., Streptococcus pneumoniae or SARS-CoV-2) into the apical (luminal) channel of the tissue model [26].
- For therapeutic intervention, add the anti-infective candidate to the perfusion medium (basolateral side) or directly to the apical surface, allowing for the study of penetration and efficacy in a physiologically relevant context.
- Endpoint Analysis:
- Transepithelial/Transendothelial Electrical Resistance (TEER): Measure in real-time to monitor barrier integrity [24].
- Effluent Collection: Analyze collected perfusate for inflammatory cytokines (e.g., IL-6, IL-8) via ELISA, and for bacterial load.
- Immunofluorescence Imaging: Fix and stain the tissues for confocal microscopy to visualize pathogen attachment, invasion, and host cell damage.
Signaling Pathways and Experimental Workflows
The transition from static models to dynamic OoCs involves integrating multiple biological and engineering principles. The diagram below outlines the key components and workflow for establishing a physiologically relevant in vitro model for anti-infective testing.
The Scientist's Toolkit: Essential Research Reagent Solutions
Building and utilizing advanced in vitro models requires a suite of specialized reagents and instruments. The following table details key components of the modern researcher's toolkit.
Table 2: Essential Materials for Organ-on-a-Chip and Advanced In Vitro Research
| Item | Function/Description | Example Use Case |
|---|---|---|
| OrganoPlate | A microfluidic 3D cell culture platform in a standard microtiter plate format (e.g., 40-, 64-, or 96-independent chips), enabling perfusion without pumps or tubing [25]. | High-throughput 3D tissue culture, barrier integrity assays, and transport studies [25]. |
| Chip-S1 (Emulate) | A PDMS-based microfluidic chip with a flexible membrane that can be subjected to cyclic stretch to mimic physiological movements like breathing or peristalsis [26]. | Lung airway and alveolus models, gut models, and studying effects of mechanical strain on cells [23]. |
| Chip-R1 (Emulate) | A rigid, non-PDMS chip designed for minimal drug absorption, making it ideal for ADME (Absorption, Distribution, Metabolism, Excretion) and toxicology studies [26]. | Accurate pharmacokinetic modeling and compound toxicity screening. |
| Extracellular Matrix (ECM) Hydrogels | Natural or synthetic hydrogels (e.g., collagen I, Matrigel) that provide a 3D scaffold to support cell growth, differentiation, and tissue morphogenesis [25] [22]. | Providing a physiological scaffold for embedding cells and forming 3D tissue structures in chips [25]. |
| Patient-Derived Organoids (PDOs) | 3D tissue cultures derived from a patient's own stem or tumor cells, retaining the genetic and phenotypic features of the original tissue [22]. | Creating personalized disease models for drug screening and studying patient-specific treatment responses [22]. |
| Transepithelial/Transendothelial Electrical Resistance (TEER) Instrument | A device to measure electrical resistance across a cellular barrier, serving as a quantitative, real-time indicator of barrier integrity and function [24]. | Assessing the formation and breakdown of biological barriers (e.g., intestinal, blood-brain barrier) in OoC models. |
The evolution from static wells to dynamic, physiologically relevant Organ-on-a-Chip models marks a paradigm shift in preclinical research. By recapitulating critical aspects of human biology—such as 3D tissue architecture, vascular perfusion, mechanical cues, and multi-organ interactions—these advanced MPS offer a powerful platform to bridge the long-standing gap between in vitro potency and in vivo efficacy [23]. For anti-infective research, this means the potential to better model host-pathogen interactions, predict clinical outcomes of antibiotic therapies, and accelerate the development of novel treatments against resistant infections. As the technology continues to standardize and scale, with the emergence of platforms like the AVA Emulation System that offers 96-chip throughput, the adoption of OoCs is poised to enhance the predictive power of drug development, reduce reliance on animal models, and usher in a new era of human-relevant biological research [26].
In the demanding landscape of drug development, particularly in oncology and anti-infective therapy, Model-Informed Drug Development (MIDD) has emerged as a transformative strategy. MIDD employs pharmacokinetic/pharmacodynamic (PK/PD) modeling and simulation to inform decision-making and optimize drug development pipelines [28]. A pivotal concept within this paradigm is the Tumor Static Concentration (TSC), a theoretical drug concentration that, if maintained constant in the plasma, results in tumor stasis—where the tumor volume neither increases nor decreases compared to the initial volume [29]. The TSC serves as a powerful, quantitative efficacy index that bridges experimental data and clinical outcomes.
The establishment of a robust In Vitro-In Vivo Correlation (IVIVC) is a primary application of TSC. IVIVC creates a predictive link between a drug's in vitro activity (e.g., cell-killing in a lab dish) and its in vivo efficacy (e.g., tumor growth inhibition in a mouse) [29]. For anti-infective drugs, the challenge is analogous: correlating in vitro microbiological activity with in vivo treatment efficacy in a complex host environment. The power of TSC lies in its ability to condense complex PK/PD relationships from both in vitro and in vivo experiments into a single, comparable metric, enabling more reliable translation from preclinical models to human clinical doses [30].
TSC Concepts and Modeling Across Therapeutic Areas
The TSC framework is versatile and can be adapted to various drug modalities, from traditional small molecules to complex biologics. The core principle involves using mathematical models to describe the system's dynamics—whether tumor cell growth or pathogen proliferation—and calculating the drug exposure level required to halt that growth.
Quantitative Foundation of TSC
The TSC is derived from the system equations of semimechanistic PK/PD models. In its fundamental form, it is the drug concentration (C) that satisfies the condition where the rate of tumor cell growth is exactly balanced by the rate of drug-induced cell kill, resulting in a net growth rate of zero:
TSC = (λ / k)
where λ represents the first-order growth rate constant of the tumor cells and k represents the second-order rate constant for the drug-induced tumor cell kill [29]. In practice, for novel compounds, these parameters (λ and k) are estimated by fitting the PK/PD model to experimental data, such as longitudinal tumor volume measurements from xenograft mouse studies [30]. The resulting TSC value provides a crucial benchmark: if the average steady-state drug concentration in plasma exceeds the TSC, tumor regression is expected; if it falls below, tumor growth is likely to continue.
Application in Antibody-Drug Conjugates (ADCs)
ADCs represent a promising but complex class of targeted cancer therapeutics. Establishing IVIVC for ADCs is critical for prioritizing lead candidates. A seminal study developed IVIVC for 19 different ADCs by calculating both an in vitro TSC (TSC~in vitro~) and an in vivo TSC (TSC~in vivo~) [29].
- TSC~in vitro~ was determined using a kinetic cell cytotoxicity assay, representing the concentration resulting in no net change in cell number compared to the start of the experiment.
- TSC~in vivo~ was determined by modeling Tumor Growth Inhibition (TGI) data from human tumor xenograft-bearing mice.
The comparison revealed a linear and positive correlation (Spearman's rank correlation coefficient = 0.82) between TSC~in vitro~ and TSC~in vivo~ across the 19 ADCs. On average, the TSC~in vivo~ was approximately 27 times higher than the TSC~in vitro~, a scaling factor that can be used to predict in vivo potency from in vitro data for new ADC candidates, thereby streamlining the selection process [29].
Translation to Clinical Dosing
The TSC concept is directly applicable to predicting human efficacious doses. A case study on RC88, a mesothelin-targeting ADC, demonstrated this translation. Researchers used three different semimechanistic PK/PD models (Simeoni, Jumbe, and Hybrid) to characterize TGI data from ovarian and lung cancer xenograft models and calculate the TSC in mice [30]. This preclinical TSC was then integrated with a prediction of human PKs, derived from a target-mediated drug disposition model built using monkey PK data, to back-calculate the required human dose expected to achieve TSC-level exposures. This integrated approach predicted an efficacious clinical dose range of 0.82 to 1.96 mg/kg administered weekly for RC88 [30].
TSC and IVIVC in Anti-Infective Drug Development
The principles of MIDD and correlation are equally critical in anti-infective drug development, which faces exciting yet challenging opportunities due to the rising threat of antimicrobial resistance [28]. The early application of MIDD at regulatory agencies involved characterizing drug molecules through population PK/PD modeling and IVIVC [28].
Table 1: Key PK/PD Indices and Correlations in Anti-Infective Development
| PK/PD Index | Definition | Role in IVIVC & MIDD |
|---|---|---|
| Minimum Inhibitory Concentration (MIC) | The lowest concentration of an antimicrobial that prevents visible growth of a microorganism. | A static, traditional endpoint used for susceptibility testing and dose stratification [28]. |
| Tumor Static Concentration (TSC) Concept | The theoretical drug exposure that results in net stasis of a pathogen population. | A dynamic, model-informed index that can integrate time-varying drug effects and host factors for superior dose optimization [28]. |
| Integrated Host-Pathogen Models | Models that incorporate host immune responses and pathogen-drug interactions. | Represents the evolving complexity in MIDD to better predict clinical outcomes for non-traditional anti-infectives [28]. |
In contrast to static PK/PD indices like the MIC, the model-informed approach embodied by the TSC concept can better describe the time-varying anti-infective effects of a drug [28]. This is crucial because the in vivo environment is dynamic, and a deeper understanding of "drug-pathogen-host" interactions is needed. For instance, the host's immune response can create microenvironments that either facilitate or impede pathogen clearance [28]. Modern MIDD frameworks are thus expanding to incorporate these host system dynamics, providing a more comprehensive basis for predicting the efficacy of novel anti-infectives, such as bacteriophages and immunomodulating agents [28].
Experimental Protocols for Establishing IVIVC
A robust IVIVC requires carefully designed and executed experiments to generate high-quality data for PK/PD modeling.
Protocol for In Vitro TSC Determination (ADC Example)
This protocol outlines the key steps for establishing an in vitro efficacy matrix [29].
- Kinetic Cell Cytotoxicity Assay: Seed cancer cells into multi-well plates and allow them to adhere.
- ADC Exposure: Treat the cells with a range of ADC concentrations. Include control wells with vehicle only.
- Longitudinal Cell Viability Measurement: Use a real-time cell analysis (RTCA) system or similar technology to monitor cell proliferation and viability kinetically over a defined period (e.g., 3-5 days), rather than at a single endpoint.
- Data Analysis and Model Fitting: Fit the resulting time-course viability data to a mathematical model that describes cell growth and ADC-induced killing. The model parameters are estimated from this data fit.
- TSC~in vitro~ Calculation: Using the fitted model, calculate the theoretical ADC concentration that would result in the final cell number being equal to the initial cell number. This concentration is the TSC~in vitro~.
Protocol for In Vivo TSC Determination (ADC Example)
This protocol describes the generation of in vivo data for PK/PD modeling [30].
- Xenograft Model Establishment: Implant human tumor cells (e.g., OVCAR-3 ovarian cancer or H292 lung cancer cells) subcutaneously into immunodeficient mice (e.g., Balb/c nude mice).
- Randomization and Dosing: Once tumor volumes reach a predetermined size (~250 mm³), randomize mice into different treatment groups. Groups typically include a vehicle control and multiple dose levels of the ADC (e.g., 0.75, 1.5, and 3 mg/kg).
- Drug Administration and PK Sampling: Administer the ADC intravenously according to the planned schedule (e.g., once weekly for 3 weeks). Collect blood samples at various time points post-dose from a subset of animals to characterize the ADC's pharmacokinetic profile.
- Tumor Volume Measurement: Measure tumor dimensions (length and width) using calipers twice weekly. Calculate tumor volume using the formula:
Volume (mm³) = 0.5 × (width²) × length. - PK/PD Modeling and TSC~in vivo~ Calculation: The longitudinal tumor volume data and corresponding PK data are co-modeled using a semimechanistic PK/PD model (e.g., Simeoni, Jumbe, or Hybrid models). The TSC~in vivo~ is derived as a secondary parameter from the fitted model, representing the constant plasma concentration that would lead to tumor stasis.
Visualization of Concepts and Workflows
Conceptual Framework of TSC and IVIVC
This diagram illustrates the core workflow of using TSC to bridge in vitro and in vivo data.
Diagram 1: TSC-based IVIVC workflow for human dose prediction.
MIDD Workflow in Anti-Infective Development
This diagram outlines the expanded MIDD workflow for anti-infectives, incorporating host-pathogen-drug interactions.
Diagram 2: MIDD framework integrating pathogen, drug, and host data.
The Scientist's Toolkit: Key Research Reagents and Materials
Table 2: Essential Reagents and Tools for TSC and IVIVC Studies
| Category | Item | Function in Experiment |
|---|---|---|
| Biological Models | Cancer Cell Lines (e.g., OVCAR-3, H292) [30] | In vitro potency screening and establishing xenograft models for in vivo efficacy studies. |
| Immunodeficient Mice (e.g., Balb/c nude) [30] | Host for human tumor xenografts to evaluate in vivo drug efficacy in a pre-clinical setting. | |
| Cynomolgus Monkeys [30] | Non-human primate model used for toxicology and translational PK studies to predict human PK. | |
| Key Reagents | Tool ADC / Drug Candidate [30] | The therapeutic molecule being evaluated, with a well-characterized Drug-Antibody Ratio (DAR) for ADCs. |
| Detection Antibodies (e.g., anti-MMAE, HRP-conjugated) [30] | Critical for developing immunoassays (ELISA) to quantify drug concentrations in biological matrices. | |
| Analytical Instruments | ELISA Plate Reader [30] | Measures drug concentration in serum/plasma samples for PK analysis. |
| Real-Time Cell Analyzer (RTCA) [29] | Enables kinetic, label-free monitoring of cell proliferation and cytotoxicity for in vitro TSC determination. | |
| Software & Models | PK/PD Modeling Software (e.g., NONMEM, Monolix, R) | Platform for building, validating, and simulating semimechanistic PK/PD models to derive TSC. |
| Semimechanistic Models (Simeoni, Jumbe, Hybrid) [30] | Pre-defined model structures that describe tumor growth and drug-induced killing, used for TSC calculation. |
The integration of Tumor Static Concentration (TSC) within a PK/PD modeling framework provides a powerful, quantitative approach to bridging in vitro potency and in vivo efficacy. This methodology enables a more rational and efficient path for drug development, from triaging lead candidates to predicting human efficacious doses. The demonstrated success of this approach in complex modalities like ADCs, coupled with its logical extension into the critical field of anti-infective research through advanced MIDD practices, underscores its transformative potential. As drug discovery confronts increasingly challenging targets and the urgent threat of antimicrobial resistance, model-informed strategies like TSC-based IVIVC will be indispensable for accelerating the delivery of novel therapies to patients.
Semi-mechanistic mathematical models represent a powerful methodology in quantitative pharmacology and therapeutics development, seamlessly integrating theoretical mechanism-based principles with empirical data-driven approaches. This review provides a comprehensive comparison of these modeling frameworks within the context of anti-infective and oncology drug development, with particular emphasis on their role in establishing robust in vitro-in vivo correlations (IVIVC). We examine fundamental model structures, experimental methodologies for parameter quantification, and implementation protocols across therapeutic domains. By systematically comparing alternative modeling approaches through structured tables and visual workflows, this guide aims to equip researchers with practical frameworks for selecting appropriate model structures based on specific research objectives, data availability, and biological complexity. The integration of these quantitative approaches provides a powerful platform for accelerating therapeutic optimization and advancing personalized medicine strategies across diverse disease areas.
Semi-mechanistic mathematical models have emerged as indispensable tools in biomedical research and therapeutic development, occupying a crucial middle ground between purely phenomenological models and fully mechanistic biological simulations [31]. These models incorporate key biological processes—such as drug exposure, pathogen/tumor growth, and treatment-induced decay—while remaining mathematically tractable for parameter estimation and prediction [32]. In anti-infective research, they provide a quantitative framework for bridging in vitro potency assessments with in vivo efficacy predictions, addressing a fundamental challenge in drug development [21].
The core strength of semi-mechanistic models lies in their ability to integrate known biology while maintaining computational feasibility. Unlike black-box models that merely describe input-output relationships, semi-mechanistic models incorporate fundamental biological principles such as tumor growth kinetics [32], antibiotic pharmacokinetic/pharmacodynamic (PK/PD) relationships [33], and immune response dynamics [31]. This balanced approach enables researchers to not only predict system behavior but also to gain insights into underlying biological mechanisms driving observed responses.
Within the context of anti-infective efficacy research, establishing robust correlations between in vitro measurements and in vivo outcomes remains a critical challenge with significant implications for drug development efficiency and clinical translation [21]. This review systematically compares semi-mechanistic modeling approaches across therapeutic domains, providing researchers with structured frameworks for model selection, implementation, and validation in preclinical and clinical settings.
Theoretical Foundations of Semi-Mechanistic Modeling
Core Mathematical Frameworks
Semi-mechanistic models typically employ differential equation systems to capture the dynamic interactions between system components. The fundamental structure integrates terms representing natural growth/decay processes with intervention-induced effects:
Ordinary Differential Equation (ODE) Systems: These represent the workhorse framework for most semi-mechanistic models, describing how system states evolve over time through rate equations [32]. In oncology, tumor growth dynamics are frequently captured using exponential, logistic, or Gompertz functions, while treatment effects are modeled through various "kill term" parameterizations [32]. For anti-infectives, microbial growth and antimicrobial-induced killing follow similar principles but with different parameter values and functional forms [33].
Key Model Components:
- System State Variables: Quantities representing biological entities (e.g., tumor volume, bacterial density, drug concentration)
- Growth Terms: Mathematical functions describing natural system expansion (e.g., exponential, logistic, or linear growth)
- Treatment Effect Terms: Functions quantifying intervention impacts (e.g., direct killing, growth inhibition)
- Transition Terms: Equations capturing conversions between system states (e.g., sensitive to resistant populations) [32]
Model Identification Approaches: Semi-mechanistic model development typically follows one of three strategies: (1) Bottom-up approaches building from first principles; (2) Top-down approaches fitting flexible functions to data; or (3) Middle-out strategies that incorporate known biology while keeping models identifiable from available data [31]. The middle-out approach has proven particularly valuable in complex domains like immuno-oncology, where some biological mechanisms are well-characterized while others remain incompletely understood [31].
Comparative Analysis of Growth and Decay Models
Table 1: Fundamental Growth Models in Biological Systems
| Model Type | Mathematical Formulation | Key Parameters | Biological Interpretation | Applications |
|---|---|---|---|---|
| Exponential Growth | dT/dt = k₉·T |
k₉: Growth rate constant | Unlimited growth proportional to current state | Early tumor growth [32]; Bacterial proliferation [33] |
| Logistic Growth | dT/dt = k₉·T·(1 - T/Tₘₐₓ) |
k₉: Growth rate; Tₘₐₓ: Carrying capacity | Density-limited growth accounting for resource constraints | Solid tumor dynamics [32] |
| Gompertz Growth | dT/dt = k₉·T·ln(Tₘₐₓ/T) |
k₉: Growth rate; Tₘₐₓ: Carrying capacity | Rapid early growth with progressive slowing | Established tumor growth with spatial constraints [32] |
| Linear Growth | dT/dt = k₉ |
k₉: Constant growth rate | Constant volume increase over time | Late-stage tumor growth [32] |
Table 2: Treatment Effect Models for Therapeutic Interventions
| Effect Model | Mathematical Formulation | Key Parameters | Mechanistic Interpretation | Therapeutic Context |
|---|---|---|---|---|
| First-Order Kill | dT/dt = f(T) - k₄·T |
k₄: Kill rate constant | Cell killing proportional to population size | Constant concentration chemotherapy [32] |
| Exposure-Dependent Kill | dT/dt = f(T) - k₄·Exposure·T |
k₄: Drug potency parameter | Killing proportional to both population and drug exposure | PK-driven dosing regimens [32] |
| Resistance-Development | dT/dt = f(T) - k₄·e^(-λ·t)·Exposure·T |
k₄: Initial kill rate; λ: Resistance emergence rate | Progressive loss of efficacy due to resistance | Long-term antimicrobial or anticancer therapy [32] |
| Emax Model | k₉' = k₉·(1 - Eₘₐₓ·Exposure/(IC₅₀ + Exposure)) |
Eₘₐₓ: Maximal effect; IC₅₀: Potency | Saturable effect following Michaelis-Menten kinetics | Targeted therapies with receptor-mediated effects [32] |
Experimental Methodologies for Model Parameterization
In Vitro Systems for Anti-Infective Potency Assessment
Establishing quantitative relationships between drug exposure and biological effect requires carefully designed experimental systems that generate data for model parameterization:
Time-Kill Kinetics Studies: These experiments characterize the temporal dynamics of antimicrobial activity by monitoring bacterial density changes over time following antibiotic exposure [27]. Unlike static endpoints like minimum inhibitory concentration (MIC), time-kill studies provide rich longitudinal data capturing both initial killing and potential regrowth due to resistance emergence or subpopulations [27]. Experimental protocols involve:
- Preparing standardized bacterial inoculums (typically 10⁵-10⁶ CFU/mL)
- Exposing cultures to fixed or dynamically changing antibiotic concentrations
- Sampling at predetermined timepoints (e.g., 0, 2, 4, 8, 24 hours) for viable counting
- Quantifying concentration-dependent killing patterns and post-antibiotic effects [27]
Hollow Fiber Infection Models (HFIM): These advanced systems bridge the gap between static in vitro assays and in vivo models by simulating human pharmacokinetic profiles against bacterial populations [27]. HFIM technology enables:
- Simulation of human drug concentration-time curves
- Prolonged observation of bacterial responses to dynamically changing concentrations
- Assessment of resistance emergence under clinically relevant exposure scenarios [27]
- Evaluation of combination therapies through simultaneous administration of multiple agents [33]
Minimum Inhibitory/Bactericidal Concentration (MIC/MBC) Determinations: While providing limited dynamic information, MIC and MBC values serve as important anchoring points for model development [27]. Standardized protocols include:
- Broth microdilution methods with standardized media and inoculum sizes
- Endpoint reading after 18-24 hours incubation
- MBC determination through subculturing from clear wells
- Quality control using reference strains [27]
Figure 1: Integrated Experimental Workflow for Semi-Mechanistic Model Development - This diagram illustrates the sequential integration of in vitro characterization, pharmacokinetic profiling, and in vivo evaluation for comprehensive model parameterization and validation.
In Vivo Systems for Model Validation
Animal models provide critical data for validating semi-mechanistic models and establishing in vitro-in vivo correlations:
Immunocompetent Tumor Models: These systems capture complex immune-tumor interactions crucial for immuno-oncology applications [31]. The TC-1/A9 cold tumor model in C57BL/6J mice exemplifies this approach, featuring:
- Subcutaneous implantation of tumor cells expressing specific antigens
- Randomized treatment assignment when tumors reach predetermined sizes
- Longitudinal tumor volume measurements
- Immunophenotyping of tumor microenvironment [31]
Infection Model Systems: Animal models of bacterial, fungal, or viral infections enable assessment of antimicrobial efficacy under physiologically relevant conditions:
- Immunocompromised models for evaluating bacteriostatic vs. bactericidal activity
- Tissue-specific infection models (e.g., pneumonia, endocarditis, meningitis)
- Monitoring of pathogen density and host response biomarkers over time
- Assessment of combination therapy efficacy and resistance suppression [33]
Protocol Considerations: Standardized experimental protocols are essential for generating high-quality data for model parameterization:
- Consistent inoculum preparation and administration routes
- Controlled dosing regimens with documented pharmacokinetics
- Frequent sampling for longitudinal assessment of response markers
- Adequate group sizes for population variability assessment [31] [33]
Implementation Protocols for Key Model Types
Tumor Growth Inhibition (TGI) Modeling
The TGI framework represents one of the most widely applied semi-mechanistic approaches in oncology:
Base Structural Model:
Where:
- T = Tumor volume
- k₉ = First-order tumor growth rate
- k₄ = Drug-induced kill rate
- λ = Rate of resistance development
- Exposure = Drug concentration driving effect [32]
Implementation Protocol:
- Data Collection: Longitudinal tumor volume measurements from control and treated cohorts
- Structural Model Selection: Exponential, logistic, or Gompertz growth functions based on control group data
- Treatment Effect Parameterization: Direct kill, cytostatic, or resistance-development models
- Parameter Estimation: Nonlinear mixed-effects modeling to estimate population and individual parameters
- Model Validation: Visual predictive checks, bootstrap analysis, and external validation [32]
Case Example: In cold tumor models, TGI models have been extended to incorporate immune activation dynamics, with successful application to combinations including antigens, TLR-3 agonists, and immune checkpoint inhibitors [31].
Integrated PK/PD Modeling for Anti-Infectives
Semi-mechanistic PK/PD models quantitatively link antibiotic exposure to microbial killing:
Structural Components:
- PK Submodel: Typically one- or two-compartment models describing plasma/tissue concentration-time profiles
- PD Submodel: Functions relating drug concentrations to microbial killing rates
- Bacterial Submodel: Equations describing bacterial growth, natural death, and drug-induced killing [33]
Implementation Protocol:
- PK Model Development: Separate characterization of drug disposition kinetics
- In Vitro PD Model: Parameterization of concentration-effect relationships using time-kill data
- Integrated PK/PD: Linking PK and PD components through driving function
- Translation to In Vivo: Incorporation of host-specific factors and immune effects
- Clinical Application: Bridging to human pharmacokinetics and infection sites [33]
Advanced Applications: Recent models incorporate bacterial subpopulations with differential susceptibility, allowing prediction of resistance emergence under various dosing scenarios [33].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Essential Research Reagents for Semi-Mechanistic Modeling Studies
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Cell Lines | TC-1/A9 tumor cells [31]; HepG2 for transfection [21]; Clinical bacterial isolates [34] | In vitro and in vivo efficacy models | Authentication, passage number, growth characteristics |
| Culture Media | Mueller-Hinton agar [34]; Sabouraud Dextrose Agar [34]; Cell-specific optimized media | Pathogen and cell line maintenance | Standardization for reproducibility across labs |
| Therapeutic Agents | Anti-PD1 [31]; TLR-3 agonists [31]; Reference antibiotics [27] | Treatment intervention studies | Source, purity, formulation stability |
| Analytical Tools | Vitek2 Compact ID System [34]; Capillary gel electrophoresis [21]; Flow cytometry | System characterization and response monitoring | Validation, sensitivity, throughput |
| Modeling Software | Monolix [31]; NONMEM; R with specialized packages | Parameter estimation and model simulation | Algorithm robustness, diagnostic capabilities |
Comparative Performance Analysis Across Therapeutic Areas
Model Predictive Performance
Table 4: Comparative Performance of Semi-Mechanistic Models Across Applications
| Model Application | Data Requirements | Prediction Accuracy | Limitations | Implementation Complexity |
|---|---|---|---|---|
| Tumor Growth Inhibition | Longitudinal tumor volume data; dosing history [32] | High for short-term predictions; moderate for long-term resistance | Limited tumor heterogeneity representation; microenvironment simplification | Moderate (requires specialized PK/PD expertise) |
| Antibacterial PK/PD | Time-kill data; population PK; resistance frequency [33] | High for efficacy prediction; moderate for resistance emergence | Often neglects host immune contributions; in vitro-in vivo translation challenges | Moderate to high (complex bacterial population dynamics) |
| Immuno-Oncology | Tumor size; immune cell counts; cytokine measurements [31] | Moderate (complex immune-tumor interactions) | High parameter uncertainty; extensive data requirements for validation | High (multiple interacting biological systems) |
| IVIVC for Vaccines | In vitro potency; animal immunogenicity [21] | Variable (depends on quality of correlate) | Limited for novel platforms; species-specific differences | Moderate (statistical correlation approaches) |
In Vitro - In Vivo Correlation Success Rates
Establishing quantitative relationships between in vitro measurements and in vivo outcomes remains a central challenge in drug development:
Vaccine Potency Correlation: For well-characterized vaccine platforms like HPV VLP vaccines, robust correlations between in vitro immunoassays and in vivo immunogenicity have been established, enabling in vitro potency assays for lot release [21]. Similar approaches are being developed for mRNA vaccines, though correlation establishment remains challenging due to additional complexity of intracellular delivery and translation [21].
Antibiotic Exposure-Response: Semi-mechanistic models have successfully linked in vitro time-kill data to in vivo efficacy in animal models, with subsequent translation to human dosing regimens [33]. Key success factors include:
- Accounting for protein binding differences between systems
- Incorporating site-specific penetration factors
- Modeling resistant subpopulation dynamics
- Considering host immune contribution to bacterial clearance [35] [33]
Oncology Translation: Predicting human efficacy from preclinical models remains challenging due to interspecies differences in drug metabolism, tumor biology, and immune function. Semi-mechanistic models improve translation through:
- Allometric scaling of pharmacokinetic parameters
- Incorporating human-specific biomarker data
- Accounting for population variability in key parameters [32]
Figure 2: Semi-Mechanistic Structure for Immuno-Oncology Response - This diagram illustrates key components and interactions in a semi-mechanistic model of immunotherapy response in cold tumors, highlighting the central role of CD8+ T-cell dynamics and resistance mechanisms [31].
Semi-mechanistic mathematical models provide a powerful quantitative framework for integrating drug exposure, biological growth, and treatment-induced decay across therapeutic domains. Through systematic comparison of model structures, experimental methodologies, and implementation protocols, this guide demonstrates how these approaches facilitate robust predictions of therapeutic efficacy and support informed decision-making in drug development.
The comparative analysis reveals that while specific mathematical forms differ between anti-infective and oncology applications, the underlying principles of integrating known biology with empirical data remain consistent. Successful implementation requires careful consideration of model purpose, data availability, and biological complexity, with middle-out approaches offering particular promise for balancing mechanistic insight with practical identifiability.
As therapeutic modalities continue to evolve, semi-mechanistic models will play an increasingly critical role in accelerating development through improved in vitro-in vivo correlations, optimized dosing strategies, and enhanced understanding of resistance mechanisms. The continued refinement and application of these quantitative frameworks represents a key frontier in model-informed drug development and personalized medicine.
In vitro-in vivo correlation (IVIVC) is defined as a predictive mathematical model describing the relationship between an in vitro property of a dosage form (usually the rate or extent of drug dissolution or release) and a relevant in vivo response (such as plasma drug concentration or amount absorbed) [36]. The establishment of a robust IVIVC has transformative implications for modern drug development, particularly for complex therapeutics like Antibody-Drug Conjugates (ADCs) and antiviral agents. For ADCs, which constitute a novel class of biopharmaceuticals designed to selectively deliver cytotoxic agents to tumor cells, IVIVC provides a mechanism to triage molecules during discovery stages, preventing unnecessary scaling-up and conserving valuable resources [29] [37]. Similarly, in the antiviral domain, Model-Informed Drug Development (MIDD) approaches that incorporate IVIVC are becoming indispensable tools for addressing the challenges posed by rapidly mutating viruses and dwindling therapeutic pipelines [28].
The fundamental value of IVIVC lies in its ability to predict in vivo performance based on in vitro data, thereby reducing the need for extensive animal studies and clinical trials while enhancing formulation optimization and quality control [9]. This review comprehensively compares and contrasts the application of IVIVC principles across two distinct therapeutic classes—ADCs in oncology and antiviral therapies—highlighting methodological frameworks, experimental data, and future directions to guide researchers and drug development professionals in leveraging these powerful correlation tools.
IVIVC for Antibody-Drug Conjugates (ADCs)
ADC Fundamentals and IVIVC Challenges
Antibody-Drug Conjugates represent a targeted approach to cancer therapy, comprising monoclonal antibodies covalently linked to cytotoxic agents via engineered chemical linkers [37]. These "magic bullets" are designed to selectively deliver potent payloads to tumor cells while minimizing damage to healthy tissues. However, ADC development faces significant challenges including antibody immunogenicity, linker instability, premature payload release, and complex pharmacokinetic-pharmacodynamic (PK/PD) relationships [38] [37]. These factors complicate the establishment of predictive IVIVC models, as the in vivo behavior of ADCs depends not only on the properties of individual components but also on their integrated performance in biological systems.
The clinical translation of ADCs necessitates balancing efficacy with toxicity, making IVIVC an invaluable tool for optimizing this therapeutic index. Currently, 15 ADCs have gained regulatory approval globally, with over 400 in various development stages [37]. For these complex molecules, IVIVC moves beyond traditional dissolution testing to encompass correlations between in vitro efficacy models and in vivo tumor growth inhibition.
Experimental Framework for ADC IVIVC
A pioneering PK/PD modeling approach for establishing IVIVC for ADC efficacy utilized nineteen different ADCs to develop correlations between in vitro and in vivo performance metrics [29]. The experimental methodology encompassed several key stages:
In Vitro Assessment: Evaluation of ADC efficacy using kinetic cell cytotoxicity assays against target cancer cells. The cytotoxicity data were characterized using a novel mathematical model to derive an in vitro efficacy parameter termed 'in vitro tumor static concentration' (TSCin vitro). TSCin vitro represents the theoretical concentration at continuous exposure that would maintain the initial cell number without increase or decrease [29].
In Vivo Evaluation: Assessment of ADC efficacy through tumor growth inhibition (TGI) studies in human tumor xenograft-bearing mice. The TGI data were characterized using a PK/PD model to derive an in vivo efficacy parameter termed 'in vivo tumor static concentration' (TSCin vivo). This represents the theoretical plasma concentration that would maintain the initial tumor volume in a mouse model [29].
Correlation Analysis: Comparison of TSCin vitro and TSCin vivo values across the 19 ADCs to establish a linear IVIVC, with a Spearman's rank correlation coefficient of 0.82 observed [29].
Table 1: Key Experimental Parameters in ADC IVIVC Development
| Parameter | Description | Application in IVIVC |
|---|---|---|
| TSCin vitro | Theoretical in vitro concentration inhibiting net cell growth | In vitro efficacy matrix derived from kinetic cell cytotoxicity assays |
| TSCin vivo | Theoretical in vivo plasma concentration inhibiting tumor growth | In vivo efficacy matrix derived from PK/PD modeling of TGI studies |
| Spearman's Correlation | Non-parametric measure of rank correlation | Quantified relationship between TSCin vitro and TSCin vivo (r=0.82) |
| Scalar Difference | Ratio between TSCin vivo and TSCin vitro | ~27-fold higher TSCin vivo versus TSCin vitro on average |
Quantitative Correlation Data and Interpretation
The study established a linear and positive IVIVC across the 19 evaluated ADCs, demonstrating that in vitro efficacy data could correctly differentiate ADCs for their in vivo efficacy [29]. The key quantitative findings included:
- A Spearman's rank correlation coefficient of 0.82, indicating a strong monotonic relationship between in vitro and in vivo potency metrics.
- On average, TSCin vivo was approximately 27 times higher than TSCin vitro, suggesting consistent differences in potency requirements between simplified in vitro systems and complex in vivo environments.
- The robustness of the correlation across multiple ADC structures supported the utility of this IVIVC approach for prioritizing ADC candidates during early development.
This correlation approach enables researchers to predict efficacious ADC concentrations in vivo using in vitro data, thereby optimizing the design of preclinical efficacy studies and accelerating the development timeline for promising candidates.
Diagram 1: ADC IVIVC Workflow illustrating the sequential process from in vitro assays to in vivo efficacy prediction.
IVIVC for Antiviral Therapies
Antiviral Development Challenges and MIDD Approaches
Antiviral drug development faces unique challenges, including the high genomic variability of viruses like HIV and influenza, which leads to rapid resistance emergence and complicates treatment strategies [28] [39]. Influenza viruses alone cause approximately one billion annual cases globally, with 290,000-650,000 deaths, highlighting the urgent need for effective therapies [39]. The error-prone RNA-dependent RNA polymerase of influenza viruses generates significant antigenic drift, while genomic segment reassortment enables antigenic shift, potentially producing pandemic strains [39].
In this context, Model-Informed Drug Development (MIDD) has emerged as a critical framework for integrating IVIVC principles into antiviral development. MIDD expands beyond traditional PK/PD modeling to incorporate host immune dynamics, viral mutation patterns, and combination therapy effects [28]. The approach allows characterization of time-varying anti-infective effects that static parameters like minimum inhibitory concentration (MIC) cannot adequately capture.
Quantitative Assessment of Antiviral Antibody Functions
Recent innovations in antiviral antibody assessment have enabled more precise IVIVC development through quantitative evaluation of functional components. For HIV treatment, researchers have developed methods comparing wild-type antibodies with Fc function-deficient mutants in animal models to quantify the contributions of different antibody functions [40]. The experimental protocol includes:
- Antibody Engineering: Creation of Fc function-deficient mutants through specific point mutations (L234F, L235E, P331S, N297A) that preserve antigen binding but eliminate effector functions.
- In Vivo Modeling: Evaluation in HIV-1 infected humanized mouse models and SHIV-infected rhesus macaque models to compare viral kinetics between wild-type and mutant antibodies.
- Mathematical Modeling: Development of precise mathematical models to quantify the relative contributions of neutralization activity versus effector functions in overall antiviral efficacy [40].
This approach has demonstrated that antibodies with intact Fc functions show significantly superior antiviral effects compared to neutralization-only variants, with earlier and faster viral load reduction observed in animal models [40].
Table 2: Clinically Approved Anti-Influenza Agents and Their Mechanisms
| Drug | Viral Target | Mechanism of Action | Administration Route | Approval Status |
|---|---|---|---|---|
| Zanamivir (Relenza) | Neuraminidase | Sialic acid analogue | Inhalation/Intravenous | FDA and EMA approved |
| Oseltamivir (Tamiflu) | Neuraminidase | Sialic acid analogue | Oral | FDA and EMA approved |
| Peramivir (Rapivab) | Neuraminidase | Sialic acid analogue | Intravenous | FDA and EMA approved |
| Baloxavir marboxil (Xofluza) | RdRp (PA) | Cap-dependent endonuclease inhibitor | Oral | FDA and EMA approved |
| Favipiravir (Avigan) | RdRp (PB1) | Nucleoside analogue | Oral | Approved only in Japan |
Integration of Host-Pathogen-Drug Interactions
A significant advancement in antiviral IVIVC is the incorporation of host system dynamics into quantitative models. Unlike traditional approaches focused primarily on drug-pathogen interactions, modern MIDD frameworks recognize the crucial role of host immune responses in determining treatment outcomes [28]. Key considerations include:
- Immune System Interactions: Host immune responses can create microenvironments that either facilitate or impede pathogen clearance, significantly influencing drug efficacy.
- Microbiome Impacts: The resident microbiota can affect drug metabolism and antiviral activity, adding another variable to correlation models.
- Genetic Predispositions: Host genetic factors influencing drug metabolism or immune function must be considered for accurate IVIVC.
This comprehensive "drug-pathogen-host" modeling approach is particularly valuable for novel antiviral modalities like monoclonal antibodies and immunomodulators, where traditional IVIVC frameworks may be insufficient [28] [40].
Diagram 2: Antiviral MIDD Framework illustrating the integration of host, pathogen, and drug factors for treatment optimization.
Comparative Analysis: ADC vs. Antiviral IVIVC Approaches
Methodological Comparison
While both ADC and antiviral IVIVC approaches share the fundamental goal of correlating in vitro data with in vivo performance, their methodological frameworks differ significantly based on therapeutic context and biological complexity.
Table 3: Methodological Comparison Between ADC and Antiviral IVIVC Approaches
| Aspect | ADC IVIVC | Antiviral IVIVC |
|---|---|---|
| Primary In Vitro Model | Kinetic cell cytotoxicity assays | Viral inhibition assays (e.g., plaque reduction) |
| Key In Vitro Parameter | Tumor Static Concentration (TSCin vitro) | Inhibition constants (IC50, EC50) |
| Primary In Vivo Model | Human tumor xenograft-bearing mice | Infected animal models (e.g., humanized mice) |
| Key In Vivo Parameter | Tumor Static Concentration (TSCin vivo) | Viral load reduction, survival benefit |
| Correlation Focus | Linking in vitro potency to tumor growth inhibition | Predicting clinical efficacy from laboratory data |
| Special Considerations | Bystander effects, target heterogeneity | Resistance emergence, host immunity |
Correlation Strength and Predictive Performance
The strength of established IVIVC varies between therapeutic classes based on model complexity and biological understanding:
- ADCs: Demonstrated strong monotonic correlations (Spearman's r=0.82) across multiple molecules, with consistent scalar relationships (27-fold difference TSCin vivo/TSCin vitro) enabling reliable prediction of in vivo efficacy from in vitro data [29].
- Antivirals: Correlation strength varies by viral target and drug class, with more robust IVIVC generally observed for direct-acting antivirals versus host-targeting agents. The integration of quantitative antibody function assessment has improved correlation robustness for antiviral monoclonal antibodies [40].
For both therapeutic classes, the level of correlation sufficient for decision-making depends on the specific application, with formulation screening requiring less robust correlations than regulatory submissions for biowaivers [9].
Essential Research Tools and Reagent Solutions
The successful establishment of IVIVC for both ADCs and antiviral therapies relies on specialized research tools and reagent systems designed to address the unique challenges of each therapeutic class.
Table 4: Essential Research Reagent Solutions for IVIVC Development
| Research Tool | Function in IVIVC | Application Context |
|---|---|---|
| Engineered ADC Mutants | Systematically vary drug-antibody ratio (DAR), linker stability, and antibody specificity | ADC optimization and mechanism studies |
| Fc Function-Deficient Antibodies | Quantify contribution of effector functions to overall antiviral activity | Antiviral antibody evaluation |
| Humanized Mouse Models | Evaluate human-specific therapeutic effects in vivo | Both ADC and antiviral development |
| PBPK Modeling Software | Simulate in vivo drug absorption, distribution, and elimination | Predictive IVIVC across therapeutic areas |
| Biorelevant Dissolution Systems | Simulate gastrointestinal conditions for oral dosage forms | Antiviral drug formulation development |
| Cytotoxicity Assay Platforms | Quantify cell killing potency in standardized formats | ADC potency assessment |
| Viral Load Quantification Assays | Precisely measure antiviral effects in vitro and in vivo | Antiviral efficacy correlation |
Future Perspectives and Concluding Remarks
The future of IVIVC for both ADCs and antiviral therapies points toward increasingly sophisticated integration of computational and experimental approaches. For ADCs, continued refinement of PK/PD models incorporating bystander effects, tumor microenvironment influences, and payload release kinetics will enhance correlation accuracy [29] [37]. The emergence of fourth-generation ADCs with optimized drug-antibody ratios and site-specific conjugation will demand corresponding advances in IVIVC methodologies to address their unique properties [37].
In the antiviral domain, MIDD approaches will increasingly incorporate host immune dynamics, viral evolution patterns, and real-world evidence to create more predictive correlation models [28]. The growing threat of multidrug-resistant pathogens necessitates IVIVC frameworks that can inform combination therapy development and address novel resistance mechanisms.
For both therapeutic classes, the convergence of artificial intelligence-driven modeling, microfluidic systems, and high-throughput screening technologies holds immense potential for augmenting the predictive power and scope of IVIVC studies [9]. These technological synergies will enable unprecedented precision in correlating in vitro data with in vivo performance, ultimately accelerating the development of next-generation biopharmaceuticals to address unmet clinical needs across oncology and infectious disease.
The consistent demonstration that robust IVIVC can successfully predict in vivo efficacy based on in vitro data underscores its transformative potential in the drug development paradigm. As these correlation approaches become more sophisticated and widely adopted, they will play an increasingly pivotal role in bridging the gap between laboratory observations and clinical outcomes, bringing us closer to the ideal of precision medicine in both oncology and virology.
Overcoming Discrepancies and Optimizing Predictive Models
In the rigorous field of anti-infective drug development, the transition from controlled laboratory experiments to living biological systems represents a formidable challenge. A therapeutic candidate demonstrating potent efficacy in a petri dish can fail utterly within a complex living organism. This divergence between in vitro (in glass) and in vivo (in living organism) results constitutes a significant bottleneck in pharmaceutical development, wasting resources and delaying the delivery of novel treatments to patients. Understanding the sources of these discrepancies is not merely an academic exercise; it is a critical necessity for improving predictive models, accelerating drug development, and effectively combating the growing threat of antimicrobial resistance [41].
The core of the problem lies in the inherent simplification of in vitro systems. While these models provide invaluable controlled conditions for initial screening, they frequently fail to capture the dynamic, multi-faceted reality of an infection within a host. Key physiological factors—host-pathogen interactions, immune responses, tissue-specific microenvironments, and pharmacokinetic variables—are often absent or inadequately represented [41] [42]. Furthermore, the growing understanding of biofilms, which are implicated in over 65% of human bacterial infections, has highlighted another major shortcoming of traditional in vitro models: their frequent reliance on planktonic (free-floating) bacteria, which behave very differently from their biofilm-embedded counterparts [41]. This article will dissect the primary sources of error leading to the in vitro to in vivo gap, providing a comparative analysis of experimental data and outlining advanced methodologies to bridge this translational divide.
Core Discrepancies: A Comparative Analysis of In Vitro and In Vivo Models
The choice between in vitro and in vivo models is not a matter of selecting a superior option, but rather of understanding their complementary strengths and limitations. In vitro studies are conducted outside a living organism, using isolated cells, microorganisms, or biomolecules in a controlled laboratory environment. Their primary advantages include cost-effectiveness, high throughput, precise control over variables, and reduced ethical concerns [43]. Conversely, in vivo studies are performed within living organisms, such as animals or humans. They offer unparalleled physiological relevance, allowing researchers to observe complex interactions between organ systems, long-term effects of interventions, and overall disease progression in a holistic context [43].
However, this physiological relevance comes at a cost. In vivo models are expensive, time-consuming, low-throughput, and raise ethical considerations regarding animal use. Moreover, interspecies differences between animal models and humans can lead to misleading results, as the organization of the immune system and pharmacokinetic profiles can vary significantly [41]. For instance, a drug's efficacy and metabolism in a mouse may not directly translate to a human patient. The following table summarizes the fundamental characteristics of each approach:
Table 1: Fundamental Comparison of In Vitro and In Vivo Models
| Feature | In Vitro Models | In Vivo Models |
|---|---|---|
| Experimental Context | Outside living organisms (e.g., cell culture, microtiter plates) [43] | Within living organisms (e.g., mice, humans) [43] |
| Physiological Relevance | Low; fails to recapitulate complex microenvironment [41] | High; provides holistic view of biological processes [43] |
| Control of Variables | High; allows for precise manipulation [43] | Low; numerous uncontrollable biological variables |
| Throughput & Cost | High throughput, cost-effective [43] | Low throughput, expensive and time-consuming [41] |
| Ethical Considerations | Minimal | Significant, especially concerning animal welfare [41] |
| Primary Role in Drug Development | Initial screening, mechanism of action studies [43] | Safety and efficacy evaluation, preclinical data for clinical trials [41] [43] |
The failure to translate in vitro success to in vivo efficacy can be attributed to several interconnected factors.
The Biofilm Challenge
Perhaps the most significant factor in chronic and device-associated infections is the biofilm mode of growth. Biofilms are structured communities of bacteria encased in a protective matrix of extracellular polymeric substances (EPS) [41]. Bacteria within a biofilm can exhibit tolerance to antibiotics at concentrations 10 to 1000 times higher than those required to kill their planktonic counterparts [41] [44]. Standard in vitro susceptibility testing, like broth microdilution, primarily uses planktonic bacteria, creating a dramatic predictive gap. The EPS matrix acts as a physical and chemical barrier, limiting antimicrobial penetration and inactivating some compounds [41]. Furthermore, biofilms harbor metabolically heterogeneous bacterial subpopulations, including dormant "persister" cells that are highly tolerant to antibiotics [44]. They can also suppress the host's innate immune response; for example, Pseudomonas aeruginosa biofilms can produce virulence factors that eliminate polymorphonuclear leukocytes (PMNs), crucial immune effector cells [41].
The Impact of Physiological Microenvironments
The culture media used in traditional in vitro assays, such as Mueller-Hinton Broth (MHB), are nutrient-rich and designed to support robust bacterial growth, but they do not mimic the conditions found in human tissues or fluids [42]. Research has demonstrated that the chemical environment (pH, ion concentration, nutrients, presence of proteins) profoundly influences bacterial physiology and, consequently, antibiotic susceptibility.
A landmark study by Heithoff et al. (2023) directly compared Minimum Inhibitory Concentration (MIC) values for antibiotics against ESKAPE pathogens in standard MHB versus physiologically representative media, including mammalian cell culture medium (DMEM), human serum, and human urine [42]. The results were striking: approximately 15% (74/504) of the MIC values obtained in physiologic media predicted a change in susceptibility that crossed a clinical breakpoint—meaning an isolate categorized as "resistant" in MHB could be "susceptible" in DMEM, or vice versa [42]. For example, ceftriaxone and piperacillin/tazobactam were effective against MRSA in DMEM but not in MHB or serum, a prediction subsequently validated in a murine sepsis model [42]. This demonstrates that standard testing can both overlook potentially effective antibiotics and recommend ineffective ones.
Pharmacokinetic and Pharmacodynamic (PK/PD) Complexity
In vitro models typically expose bacteria to a static concentration of an antimicrobial. In vivo, however, drug concentrations are dynamic—they rise and fall over time due to absorption, distribution, metabolism, and excretion (ADME) [45]. The successful in vivo activity of an antibiotic depends on achieving a concentration at the site of infection that is sufficient to inhibit or kill the pathogen for a necessary duration, a relationship described by PK/PD indices. Furthermore, the presence of the drug at sub-therapeutic concentrations in certain tissues can inadvertently promote the development of resistance.
The Host Immune System
A critical component entirely missing from standard in vitro assays is the host's immune system. In vivo, an anti-infective agent does not work in isolation; it often functions synergistically with immune mechanisms such as antimicrobial peptides, complement proteins, and phagocytic cells like neutrophils and macrophages [42] [46]. A drug that appears merely inhibitory in vitro might be decisively bactericidal in vivo due to this cooperative effect with the immune system. Conversely, some pathogens can evade or suppress immune responses in ways that cannot be modeled in a test tube.
Case Studies and Supporting Data
Case Study 1: Generic Oxacillin Efficacy
A compelling example of the in vitro-in vivo gap was demonstrated in a study comparing 11 generic oxacillin products with the innovator product [45]. The study assessed pharmaceutical equivalence, MIC/MBC (Minimum Bactericidal Concentration), and efficacy in a neutropenic mouse thigh infection model.
Table 2: Discrepancy in Generic Oxacillin Evaluations [45]
| Test Category | Findings | Implied Conclusion by Standard Regulation |
|---|---|---|
| Pharmaceutical Equivalence | 4 of 11 generics failed due to significant differences in potency. | Pharmaceutical equivalence is assumed to predict therapeutic equivalence. |
| In Vitro Activity (MIC/MBC) | All products, including pharmaceutically non-equivalent ones, were indistinguishable from the innovator. | In vitro activity is assumed to predict in vivo efficacy. |
| In Vivo Efficacy (Mouse Model) | All generics failed therapeutic equivalence, showing lower maximum effect (Emax) and requiring higher doses for effect. | Conclusion: Pharmaceutical or in vitro equivalence does not guarantee therapeutic equivalence. |
This study underscores a critical flaw in regulatory logic: the assumption that pharmaceutical equivalence and standard in vitro testing are sufficient predictors of clinical performance. The complex in vivo environment revealed deficiencies in the generic products that simple in vitro tests could not detect [45].
Case Study 2: Physiological Media Improves Predictive Accuracy
The research by Heithoff et al. provides quantitative data supporting the reform of antimicrobial susceptibility testing (AST) standards [42]. Their head-to-head comparison of AST in MHB versus physiological media (DMEM, serum, urine) against ESKAPE pathogens yielded clinically significant discrepancies.
Table 3: MIC Changes in Physiologic Media vs. Standard MHB [42]
| Pathogen | Antibiotic | MIC in MHB (μg/mL) | MIC in DMEM (μg/mL) | Susceptibility Change | In Vivo Validation (Mouse Sepsis) |
|---|---|---|---|---|---|
| MRSA USA300 | Ceftriaxone | 256 (R) | 8 (S) | R → S | 10/10 survivors |
| MRSA USA300 | Piperacillin/Tazobactam | 256 (R) | 8 (S) | R → S | Effective |
| A. baumannii | Colistin | 0.5 (S) | 4 (R) | S → R | Not Effective |
This data powerfully argues that testing in physiologically relevant media significantly increases the diagnostic accuracy of AST, potentially salvaging useful existing antibiotics and improving the success rate of new drug discovery [42].
Methodologies for Enhanced Prediction
To bridge the translational gap, researchers are developing more sophisticated models that incorporate greater physiological relevance.
- Advanced In Vitro Biofilm Models: Moving beyond static plate assays, models like the Calgary Biofilm Device, flow cells, and drip-flow reactors allow for the formation of mature biofilms under relevant fluid shear conditions, providing a more realistic platform for testing anti-biofilm agents [44].
- Ex Vivo and Tissue-Engineered Models: Using explanted tissues (e.g., pig skin) or engineered human tissue constructs (ex vivo) can maintain some native tissue architecture and function, offering an intermediate model between simple in vitro and complex in vivo systems [47].
- Organ-on-a-Chip Technology: These microfluidic devices culture living cells in channels that mimic the structure and function of human organs, allowing for the incorporation of fluid flow, mechanical forces, and even multi-organ interactions. They represent a cutting-edge approach for studying host-pathogen interactions and drug efficacy in a highly controlled yet physiologically relevant context [41].
- Integrated Testing Cascades: A robust preclinical assessment involves a cascade of models. For example, the antimicrobial peptide DPK-060 was sequentially evaluated in vitro (showing MMC <5 μg/mL), then in an ex vivo pig skin wound model (≥99% reduction), and finally in an in vivo mouse surgical site infection model (≥94% reduction), confirming its efficacy across systems and de-risking its further development [47].
The following diagram illustrates the logical relationship between the sources of divergence and the advanced models designed to address them.
The Scientist's Toolkit: Essential Research Reagent Solutions
To implement the methodologies discussed, researchers rely on a suite of specialized reagents and tools. The table below details key solutions for improving the physiological relevance of anti-infective testing.
Table 4: Key Research Reagent Solutions for Enhanced Infection Modeling
| Reagent / Tool | Function & Rationale | Example Application |
|---|---|---|
| Physiological Culture Media | Replaces nutrient-rich broths; mimics chemical environment (ions, pH, proteins) of host tissues to elicit clinically relevant bacterial physiology [42]. | Dulbecco's Modified Eagle Medium (DMEM); pooled human serum or urine for AST [42]. |
| Standardized Biofilm Inoculum | Provides a consistent, high-density starting population of biofilm-grown bacteria for susceptibility testing, crucial for evaluating anti-biofilm agents. | Preparation of biofilm coupons from models like the MBEC (Minimum Biofilm Eradication Concentration) assay [44]. |
| Extracellular Matrix (ECM) Components | Used to create more realistic 3D cell culture environments that better mimic human tissue architecture and cell-matrix interactions. | Collagen, fibrin, or Matrigel for constructing ex vivo or organ-on-a-chip infection models. |
| In Situ Gelling Systems | Provides a delivery vehicle that can maintain a hydrated environment and allow for sustained release of antimicrobials, useful for topical application testing. | Poloxamer gels used in ex vivo and in vivo models for topical peptide delivery [47]. |
| Specialized Animal Model Reagents | Enables the creation of specific, reproducible infection models in laboratory animals for final preclinical validation. | Immunosuppressants like cyclophosphamide to create neutropenic mouse models for studying infection progression [45]. |
The divergence between in vitro and in vivo results in anti-infective research is not an insurmountable barrier but a call for more sophisticated and physiologically relevant approaches. The primary sources of error—inadequate modeling of biofilms, non-physiological culture conditions, PK/PD complexity, and the absent host immune response—are now clearly identified. As evidenced by the compelling data from comparative studies, the research community is developing powerful solutions. The integration of physiological media, advanced biofilm models, human cell-based systems like organs-on-chips, and rigorous ex vivo and in vivo validation cascades represents the path forward. By systematically addressing these sources of error, researchers can enhance the predictive power of preclinical studies, accelerate the development of effective new therapies, and ultimately improve the success rate of translating laboratory discoveries into clinical solutions for combating infectious diseases.
In the field of anti-infective efficacy research, a fundamental disconnect persists between standard antimicrobial susceptibility testing (AST) and the clinical reality of bacterial infections. Conventional AST methods, including minimum inhibitory concentration (MIC) determinations, are performed on planktonic (free-floating) bacterial cells, yet an estimated 65-80% of all infections are considered biofilm-related [48]. The biofilm lifestyle, characterized by surface-attached communities of microorganisms embedded in an extracellular polymeric matrix, confers a tremendous impact on antibiotic susceptibility that standard planktonic-based AST fails to capture [48]. This discrepancy creates a critical gap in predicting treatment outcomes, particularly for device-related infections and chronic conditions such as cystic fibrosis, osteomyelitis, and chronic wounds.
The transition from planktonic to biofilm growth involves a phenotypic shift that dramatically reduces antimicrobial susceptibility through multiple mechanisms, including physiological heterogeneity, reduced metabolic activity, and physical barrier function [49] [44]. This review objectively compares the performance of biofilm-specific assays against traditional planktonic methods within the broader thesis of in vitro versus in vivo correlation of anti-infective efficacy. By examining experimental data and methodologies, we provide researchers and drug development professionals with a framework for evaluating these tools in the context of preclinical antimicrobial validation.
Understanding Biofilm Complexity and Its Therapeutic Implications
Biofilm Lifecycle and Architecture
The biofilm lifecycle progresses through four distinct phases: (A) initial attachment of planktonic cells to a surface, (B) early development with microcolony formation, (C) maturation into a complex three-dimensional structure, and (D) controlled detachment and dispersal [49]. This developmental process creates a highly heterogeneous environment with gradients of nutrients, oxygen, and metabolic waste products that significantly influence bacterial physiology and antibiotic susceptibility [48] [49].
The extracellular polymeric substance (EPS) matrix may account for 50% to 90% of the biofilm's total biomass, with composition varying by bacterial strain, environmental conditions, and biofilm age [49]. This matrix acts as a physical barrier to antibiotic penetration while simultaneously housing microbial communities with diverse metabolic states, including dormant "persister cells" that exhibit exceptional tolerance to antimicrobial agents [49].
Mechanisms of Biofilm-Associated Antimicrobial Tolerance
Biofilms employ multiple concurrent strategies to evade antimicrobial killing, including:
- Physical Barrier Function: The EPS matrix limits antibiotic penetration through binding and sequestration [44].
- Metabolic Heterogeneity: Gradients of nutrients and oxygen create microenvironments with variable metabolic activity, including dormant persister cells [49] [44].
- Altered Microenvironment: Local conditions can neutralize antibiotic activity (e.g., β-lactams degraded by pH changes) [48].
- Adaptive Stress Responses: Biofilm cells activate general stress response pathways that increase tolerance [48].
- Efflux Pump Regulation: Biofilm-specific efflux systems are upregulated, enhancing antibiotic extrusion [48].
These mechanisms collectively contribute to biofilm tolerance levels that can be 100 to 1000 times higher than those required to eradicate their planktonic counterparts [50] [44].
Comparative Analysis of Susceptibility Testing Methods
Traditional Planktonic Susceptibility Testing (MIC)
The minimum inhibitory concentration (MIC) assay represents the current gold standard for antimicrobial susceptibility testing in clinical microbiology laboratories. This method determines the lowest concentration of an antimicrobial agent that prevents visible growth of planktonic bacteria in a growth medium [50]. MIC values are compared against established breakpoints from organizations like EUCAST and CLSI to categorize isolates as susceptible, intermediate, or resistant [48].
While MIC testing provides valuable data for planktonic infections, it demonstrates poor correlation with treatment outcomes for biofilm-associated infections [48] [51]. This limitation stems from fundamental physiological differences between planktonic and biofilm-grown bacteria, as biofilm cells express distinct genetic profiles that significantly alter their susceptibility profiles [48].
Biofilm-Specific Susceptibility Testing (MBEC)
The minimum biofilm eradication concentration (MBEC) assay measures the concentration of an antimicrobial required to eradicate bacteria within an established biofilm [50]. Unlike MIC testing, MBEC assays account for the multicellular, matrix-embedded nature of biofilms, providing a more clinically relevant measure of antibiotic efficacy for biofilm infections.
MBEC assays typically involve growing biofilms on abiotic surfaces (such as polystyrene pegs or implant materials), exposing them to antimicrobial agents, and then determining bacterial viability after treatment [50]. The resulting MBEC values are generally significantly higher than MIC values for the same bacterium-antibiotic combination, reflecting the enhanced tolerance of biofilm-grown bacteria.
Table 1: Comparative MIC and MBEC Values for Staphylococcus aureus Strains (Data from [50])
| Antibiotic | Bacterial Strain | MIC (μg/mL) | In Vitro MBEC (μg/mL) | In Vivo MBEC (μg/mL) | Fold Increase (MBEC/MIC) |
|---|---|---|---|---|---|
| Gentamicin | MSSA (UAMS-1) | 0.25-0.5 | 256-1024 | 2048->4096 | 1024-8192 |
| Gentamicin | MRSA (USA300LAC) | 0.5-1 | 256-1024 | 2048->4096 | 512-4096 |
| Vancomycin | MSSA (UAMS-1) | 1-2 | 2048-4096 | >4096 | 1024-2048 |
| Vancomycin | MRSA (USA300LAC) | 1-2 | 2048-4096 | >4096 | 1024-2048 |
| Cefazolin | MSSA (UAMS-1) | 0.25-0.5 | 2048-4096 | >4096 | 4096-8192 |
In Vitro versus In Vivo Correlation in Anti-infective Efficacy Research
Methodological Considerations in Biofilm Assays
A critical challenge in biofilm research is the significant method-dependence of efficacy measurements. A statistical meta-analysis of published data on antimicrobial efficacy against biofilms revealed that the particular experimental method used is the most important factor determining test outcome [52]. While dose-response relationships (greater killing with higher doses or longer treatment times) were consistently observed within individual studies using identical methods, these relationships disappeared when data from multiple studies using diverse methods were pooled [52].
Key methodological factors influencing antibiofilm efficacy measurements include:
- Surface area/volume ratio of the biofilm growth system
- Areal biofilm cell density at time of testing
- Biofilm maturation time before antibiotic exposure
- Nature of the substratum on which biofilms form
- Composition of the growth medium and flow conditions
Table 2: Impact of Methodological Variables on Biofilm Efficacy Measurements (Data from [52])
| Methodological Variable | Impact on Efficacy Measurement | Recommended Reporting Standards |
|---|---|---|
| Surface Area/Volume Ratio | Influences antimicrobial penetration and contact | Report specific ratios for test systems |
| Areal Biofilm Cell Density | Affects inoculum effect and killing kinetics | Quantify density at time of treatment |
| Biofilm Maturation Time | Alters matrix composition and tolerance | Standardize maturation times within studies |
| Substratum Material | Impacts attachment and biofilm architecture | Use clinically relevant materials when possible |
| Antimicrobial Exposure Conditions | Affects dose-response relationships | Include benchmark agents for comparison |
Advanced In Vitro Biofilm Models
Innovative in vitro approaches have been developed to better mimic the complexity of in vivo biofilms:
- Microfluidic Systems: These devices incorporate flow conditions that create nutrient and oxygen gradients similar to those found in clinical biofilms, supporting the development of more relevant architectural features [49].
- 3D-Printed Models: Advanced manufacturing techniques allow creation of complex structures that better simulate anatomical features and support more realistic biofilm development [49].
- Co-culture Systems: Microcosm models that include human cells or host matrix components enable study of host-pathogen interactions during antibiotic treatment [49].
- Multi-Species Biofilms: Models incorporating multiple bacterial species better represent clinical scenarios where interspecies interactions alter antimicrobial susceptibility [53].
In Vivo Validation of Biofilm Assays
The translational value of in vitro biofilm models must be validated through in vivo studies. Research comparing in vitro and in vivo models of device-related infection demonstrated that results between the two models correlated well, with correlation coefficients of 0.85-0.96 for suspended bacteria and 0.72-0.97 for adherent bacteria across different staphylococcal species [51]. This correlation supports the use of well-designed in vitro systems as predictive tools for in vivo efficacy.
A novel MBEC assay using in vivo biofilms formed on orthopedic implants in a rodent model revealed that in vivo MBEC values were substantially higher than those obtained from in vitro biofilms [50]. For instance, while in vitro MBEC values for gentamicin against Staphylococcus aureus biofilms ranged from 256-1024 μg/mL, the in vivo implant MBEC values ranged from 2048 μg/mL to more than 4096 μg/mL [50]. This discrepancy highlights the importance of host factors and the complex in vivo microenvironment in enhancing biofilm tolerance.
Experimental Protocols for Biofilm Susceptibility Testing
Standard MBEC Assay Protocol for Orthopedic Implants
The following protocol adapts the MBEC assay for evaluating antibiotic efficacy against biofilms formed on orthopedic implants, based on methodology from [50]:
- Biofilm Formation: Sterilized stainless-steel screws are incubated in tryptic soy broth with the test microorganism for 24-72 hours with shaking at 150 rpm to allow biofilm development.
- Biofilm Maturation: Implants with established biofilms are washed twice with phosphate-buffered saline to remove non-adherent bacteria.
- Antibiotic Exposure: Biofilm-coated implants are transferred to microtiter plates containing serial dilutions of antimicrobial agents and incubated for 24 hours with shaking.
- Viability Assessment: After antibiotic exposure, implants are transferred to tubes containing fresh medium, and biofilms are disaggregated using the Vortex-Sonication-Vortex Method (VSVM).
- Bacterial Recovery: The resulting suspensions are replated and incubated to determine bacterial viability, with MBEC defined as the lowest antibiotic concentration that prevents biofilm recovery.
In Vivo MBEC Assay Protocol
For measurement of MBEC against in vivo formed biofilms [50]:
- Animal Infection Model: Rats are implanted with contaminated stainless steel screws in the femoral canal.
- Biofilm Development: Animals are euthanized at specified timepoints (e.g., days 3 and 14) to represent immature and mature biofilms.
- Implant Harvesting: Explanted devices are washed gently to remove non-adherent cells while preserving the biofilm.
- Ex Vivo Antibiotic Challenge: Implants are incubated with various antibiotic concentrations for 24 hours.
- Viability Assessment: Bacterial recovery is determined using the VSVM method and CFU enumeration, with in vivo MBEC and in vivo implant MBEC calculated separately.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for Biofilm Susceptibility Testing
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| Polystyrene Peg Lids | High-throughput biofilm formation for MBEC assays | MBEC Assay System |
| Medical-Grade Implant Materials | Clinically relevant substratum for biofilm growth | Stainless steel screws, titanium alloy coupons |
| Tryptic Soy Broth (TSB) | Standardized growth medium for biofilm development | With/without glucose supplementation |
| Enzymatic Matrix Disruption Agents | Breakdown of EPS for bacterial recovery | DNase I, dispersin B, proteinase K |
| SYTOX Green Stain | Assessment of membrane permeability in biofilm cells | Fluorescence indicates compromised membranes |
| Crystal Violet | Total biofilm biomass quantification | Absorbance measurement at 590 nm |
| Microfluidic Flow Cells | Biofilm growth under controlled shear stress | With confocal microscopy compatibility |
| Neutralization Buffers | Validation of antimicrobial efficacy testing | Dey-Engley neutralization medium |
The incorporation of biofilm-specific assays such as MBEC testing represents a critical advancement in anti-infective efficacy research. The data clearly demonstrate that conventional planktonic AST fails to predict antibiotic efficacy against biofilm-associated infections, creating a translational gap in therapeutic development. While methodological standardization remains a challenge, biofilm-specific assays show significantly improved correlation with in vivo outcomes for device-related and chronic infections.
For researchers and drug development professionals, the integration of biofilm susceptibility testing at multiple stages of the development pipeline is essential. This includes employing both standardized methods and clinically relevant research models that account for the complexities of in vivo biofilms [52]. The continued refinement of these assays, coupled with advanced modeling approaches that better simulate the host environment, will enhance our ability to predict clinical efficacy and develop more effective therapies for the persistent challenge of biofilm-associated infections.
The rise of antimicrobial resistance and the challenges in treating complex diseases like cancer have positioned combination therapy as a cornerstone of modern therapeutic strategy. This approach involves using two or more therapeutic agents simultaneously to achieve a combined effect greater than the sum of their individual effects—a phenomenon known as synergy [54]. The fundamental rationale stems from the limitations of monotherapies, which often yield insufficient responses or encounter rapid development of treatment resistance [54] [55]. In oncology, for instance, monotherapies are frequently limited by the development of drug resistance to chemo-, targeted-, or immunotherapies [56]. Similarly, in anti-infective therapy, conventional antibiotics are progressively losing effectiveness against multidrug-resistant (MDR) pathogens such as the WHO ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.) [54].
Combination therapy addresses these challenges through multiple mechanistic strategies. It can enhance efficacy while potentially reducing antibiotic dosage and minimizing side effects [54]. In cancer treatment, particularly with immune checkpoint inhibitors (ICIs), combination strategies work through multiple interconnected mechanisms such as enhancing tumor immunogenicity, improving neoantigen processing and presentation, and augmenting T-cell infiltration and cytotoxic potentials [57]. The ultimate goal is to overcome resistance mechanisms that render monotherapies ineffective, whether through pharmacological synergism, targeting multiple pathways simultaneously, or preventing the emergence of resistant subpopulations.
In Vitro and In Vivo Models: Methods for Evaluating Combination Therapies
Experimental Protocols for Synergy Assessment
Time-Kill Kinetics Assay: This method evaluates the temporal dynamics of antibacterial activity by assessing how sterilization levels change over time following antibiotic administration [27]. Unlike static methods such as MIC, which measure bacterial inhibition after overnight exposure, time-kill studies track bacterial count reduction over varying time intervals, providing a dynamic perspective on drug effects [27]. This protocol is frequently employed to ascertain whether synergistic effects exist when antibiotics are used in combination. In practice, bacterial cultures are exposed to antibiotics alone and in combination, with samples collected at predetermined time points (e.g., 0, 3, 6, 12, 24 hours), serially diluted, and plated on agar to quantify viable colonies. Synergy is demonstrated when the combination reduces bacterial counts by ≥2-log10 compared to the most active single agent.
Checkboard Dilution Method: This technique systematically tests multiple concentration combinations of two antimicrobials to calculate the Fractional Inhibitory Concentration Index (FICI). Using 96-well microtiter plates, researchers create two-fold serial dilutions of both drugs in intersecting gradients, inoculate wells with a standardized microbial suspension, and determine MIC values for each drug alone and in combination after incubation. The FICI is calculated as (MIC of drug A in combination/MIC of drug A alone) + (MIC of drug B in combination/MIC of drug B alone), where FICI ≤0.5 indicates synergy, 0.5-4.0 indicates additivity/indifference, and >4.0 indicates antagonism.
Post-Antibiotic Effect (PAE) Studies: PAE measures the persistent suppression of bacterial growth after brief antibiotic exposure, which has implications for dosing intervals in combination regimens [27]. The experimental protocol involves exposing logarithmic-phase bacteria to antibiotics for a short period (typically 1-2 hours), removing the drugs by washing, dilution, or antibiotic inactivation, and then monitoring bacterial regrowth by measuring turbidity or viable counts over time. The PAE is calculated as PAE = T - C, where T is the time required for treated cultures to increase 1-log10 above the count immediately after drug removal, and C is the corresponding time for untreated controls. Combining antibiotics with different PAE profiles can optimize dosing schedules and enhance bacterial suppression.
Advanced Computational and Statistical Approaches
Machine Learning Prediction Models: Computational approaches help prioritize promising combinations from the vast number of possibilities. One method uses random forest models trained on single-drug efficacy data (represented as GI50 values) to predict synergistic combinations without detailed mechanistic understanding [55]. Features include the mean and difference of single-agent dose responses across cell lines, with models achieving significant predictive power (AUC = 0.866 for synergy prediction in mutant BRAF melanoma) [55]. This approach allows systematic in-silico screening before experimental validation.
SynergyLMM Framework: For in vivo studies, SynergyLMM provides a comprehensive statistical framework based on linear mixed models that account for inter-animal heterogeneity and longitudinal tumor growth measurements [56]. This method supports various synergy models (Bliss, HSA, Response Additivity) and offers time-resolved synergy scores with uncertainty quantification, addressing limitations of endpoint-based analyses [56]. The workflow includes data normalization, model fitting, statistical diagnosis, synergy assessment, and power analysis—implemented through both R package and web application for accessibility.
The following diagram illustrates the integrated experimental-computational workflow for evaluating combination therapies:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 1: Key Research Reagents for Combination Therapy Studies
| Reagent/Material | Function/Application | Examples/Notes |
|---|---|---|
| Cell Line Panels | Models for high-throughput drug screening | Cancer cell lines (e.g., GDSC database); ESKAPE pathogen isolates [54] [58] |
| Hollow Fiber Infection Model (HFIM) | In vitro system simulating human PK parameters | Enables prolonged antibiotic exposure studies; bridges in vitro and in vivo testing [27] |
| Animal Disease Models | In vivo efficacy and toxicity assessment | Mouse models (e.g., patient-derived xenografts); murine infection models [56] |
| Synergy Scoring Algorithms | Quantifying drug interaction effects | Bliss independence, Loewe additivity, HSA, ZIP models [56] [58] |
| Pharmacokinetic/Pharmacodynamic (PK/PD) Modeling Software | Predicting drug exposure-response relationships | Critical for translating in vitro findings to clinical dosing regimens [27] |
Quantitative Comparison of Combination Therapy Outcomes
Anti-Infective Combination Therapies
Table 2: Clinical Outcomes of Anti-Infective Combination Therapies vs. Monotherapy
| Infection Type / Pathogen | Therapeutic Regimen | Clinical Success Rate | Key Findings |
|---|---|---|---|
| Carbapenem-ResistantPseudomonas aeruginosa | Combination TherapyMonotherapy | 73.1% [59]60.4% [59] | Combination therapy significantly correlated with clinical success (OR, 0.559, 95% CI, 0.321-0.976; p=0.041) [59] |
| Carbapenem-ResistantGram-Negative Bacteria | Tigecycline +Meropenem/Imipenem/Cefoperazone-Sulbactam | 80% (32/40 patients)Good clinical response [54] | Resensitizes resistant bacteria to carbapenems; No serious adverse events, though increased liver enzymes noted [54] |
| Helicobacter pylori | L. brevis + Vitamin D3 | Significant reduction inH. pylori adhesion [54] | Anti-inflammatory and anti-oxidative effects; Potential complementary therapeutic strategy [54] |
Oncology Combination Therapies
Table 3: Efficacy of Cancer Combination Therapies in Clinical Settings
| Cancer Type | Therapeutic Regimen | Clinical Outcomes | FDA Approval Status |
|---|---|---|---|
| Non-Small Cell Lung Cancer(non-squamous) | Pembrolizumab +Carboplatin/Pemetrexed | Enhanced response rate and PFS vs chemotherapy alone [57] | Approved as first-line treatment (2018) [57] |
| Triple-Negable Breast Cancer | Atezolizumab +nab-Paclitaxel | Significant improvement in OS and PFS vs chemotherapy alone [57] | Approved for unresectable locally advanced or metastatic TNBC [57] |
| Advanced Melanoma | Local Chemotherapy +anti-CTLA-4 | Improved response rate and PFS through enhanced T-cell infiltration [57] | Demonstrated in clinical studies [57] |
Correlation Between In Vitro and In Vivo Findings: Key Challenges
The translation of in vitro combination synergy to clinical efficacy faces several significant hurdles that researchers must acknowledge and address.
Host Factor Omission: In vitro models inherently lack critical elements of the host environment, including immune responses, pH variations, iron abundance, and other dynamic conditions at anatomic infection sites [60]. These factors substantially influence antibiotic activity but are not replicated in plate-based assays. Similarly, tumor microenvironment complexities, including heterogeneous cell populations and stromal interactions, are poorly captured in monolayer cell cultures.
Pharmacokinetic/Pharmacodynamic (PK/PD) Complexity: Drug combinations that show promise in vitro may fail in vivo due to divergent tissue penetration profiles, protein binding characteristics, or metabolism pathways [60] [27]. A drug might penetrate effectively to the infection site while its combination partner does not, eliminating any potential synergy that was observed in vitro. This underscores why a low MIC does not always correlate with clinical efficacy, as antibiotic distribution varies significantly across different tissue types [27].
Bacterial Phenomena Not Fully Captured In Vitro: The development of biofilms and bacterial escape mechanisms such as persistence and tolerance may require more time than allotted for standard in vitro experiments [60]. These phenomena significantly impact treatment outcomes but are challenging to replicate in short-term laboratory assays.
Clinical Trial Evidence Gaps: Well-powered randomized clinical trials have often failed to confirm benefits suggested by in vitro synergy studies. The AIDA trial showed no benefit in adding meropenem to colistin for severe carbapenem-resistant Gram-negative infections, while the CAMERA2 trial found no benefit in adding an anti-staphylococcal beta-lactam to vancomycin or daptomycin for staphylococcal bloodstream infections—indeed, this trial was stopped early due to increased kidney injury in the combination group [60].
The relationship between experimental models and clinical translation can be visualized as follows:
Combination therapies represent a pivotal shift in combating antimicrobial resistance and improving cancer treatment outcomes. While promising in vitro synergy data continue to emerge, the translation to clinical success requires careful consideration of host factors, PK/PD complexities, and appropriate clinical trial design [60] [61]. Future research should prioritize advanced in vivo models that better recapitulate human disease, refined computational approaches for synergy prediction, and clinical trials specifically designed to identify patient subgroups most likely to benefit from combination approaches [56] [58] [62].
The integration of mechanistic studies with robust clinical validation will be essential to fully harness the potential of combination therapies. As noted in recent research, "investing in their development and clinical integration is not merely an option but a critical necessity to protect the foundation of effective antimicrobial therapy" [54]. For both infectious diseases and oncology, the strategic combination of therapeutic agents, guided by rigorous preclinical evidence and thoughtful clinical study design, offers a promising path forward against the mounting challenge of treatment resistance.
The efficacy and safety of novel anti-infective therapies are fundamentally dependent on the predictive accuracy of the biological models used in preclinical testing. Research consistently reveals a significant disconnect between traditional in vitro assays and clinical outcomes, largely attributable to their failure to replicate complex in vivo conditions. This discrepancy poses a substantial challenge in pharmaceutical development, particularly for complex infections involving biofilms and multi-drug resistant organisms. Within this context, advanced animal model systems have emerged as indispensable tools for bridging the translational gap. This guide provides a comparative analysis of two sophisticated modeling approaches: ultrasonic atomization infection models and humanized mouse systems. The integration of these optimized models into preclinical pipelines significantly enhances the correlation between experimental data and clinical efficacy, thereby de-risking drug development and accelerating the delivery of novel anti-infectives to patients.
Ultrasonic Atomization Infection Models
Ultrasonic atomization is an advanced methodology for establishing highly consistent and physiologically relevant pulmonary infection models in laboratory animals. This technique employs an ultrasonic nebulizer to generate a fine, inhalable aerosol of a bacterial suspension, enabling the uniform deposition of pathogens deep within the murine respiratory tract. The core procedure involves culturing the target pathogen, such as multi-drug resistant Acinetobacter baumannii, to a standardized concentration (e.g., 0.5 McFarland standard). The bacterial suspension is then placed into the nebulizer reservoir, and animals are exposed to the aerosol within a contained chamber for a predetermined duration. This process results in a robust and reproducible infection, effectively mimicking natural acquisition via inhalation [63].
A key application of this model is for the in vivo evaluation of antibiotic efficacy. The standard experimental workflow, used to assess combinations like minocycline with amikacin or rifampicin, is as follows [63]:
- Infection Establishment: Mice are infected with multi-drug resistant A. baumannii via ultrasonic atomization.
- Treatment Initiation: Therapy is commenced 24 hours post-infection.
- Efficacy Assessment: Animals are monitored for mortality over a 7-day period. Additionally, bacterial load and inflammatory response in lung tissues are quantified post-sacrifice to determine the therapeutic outcome.
Quantitative Efficacy Data from an MDRA. baumanniiModel
The translational value of the ultrasonic atomization model is demonstrated by its ability to generate clear, comparative efficacy data for different therapeutic regimens. The table below summarizes findings from a study evaluating single-agent and combination therapies against a multi-drug resistant A. baumannii lung infection [63].
Table 1: In Vivo Efficacy of Antibiotic Regimens in an Ultrasonic Atomization Mouse Model of MDR A. baumannii Pneumonia
| Treatment Group | Mortality Rate | White Blood Cell (WBC) Count (x10⁹/L) | Lung Histopathology Findings |
|---|---|---|---|
| Model Control (Untreated) | 100% | Not Reported | Severe inflammation, vasodilation, congestion, and hemorrhage by 48 hours. |
| Tigecycline (TIG) | Data Not Specified | Lower than combination groups | Inflammation gradually recovered with clear structures after 3 days of therapy. |
| Polymyxin B (PB) | Data Not Specified | Lower than combination groups | Inflammation gradually recovered with clear structures after 3 days of therapy. |
| Minocycline + Amikacin (MNO+AMK) | Significantly Lower | Higher than TIG and PB groups | Inflammation gradually recovered with clear structures after 3 days of therapy. |
| Minocycline + Rifampicin (MNO+RIF) | Significantly Lower | Higher than TIG and PB groups | Inflammation gradually recovered with clear structures after 3 days of therapy. |
The data reveal that combination therapies, particularly minocycline-based regimens, demonstrated superior in vivo efficacy compared to single-agent therapy, evidenced by significantly lower mortality rates and a more robust immune cell recruitment (higher WBC counts) [63]. This model successfully identified synergistic drug interactions that were consistent with in vitro findings, thereby validating its utility for preclinical drug screening.
Humanized Mouse Systems
Humanized mouse models are sophisticated in vivo platforms generated by engrafting human tissues or cells into immunodeficient mice, thereby creating a chimeric system capable of modeling human-specific immune responses and pathogen interactions. These models are foundational for studying human-tropic infectious diseases and immuno-oncology. The creation of these models relies on mouse strains with severe combined immunodeficiency, such as those carrying mutations in the IL-2 receptor common gamma chain (IL-2rγnull), which profoundly impairs innate and adaptive immunity and allows for superior engraftment of human cells [64].
The three primary approaches for generating humanized mice are [64]:
- Hu-PBL-SCID: Model is generated by injecting human peripheral blood leukocytes (PBL) into immunodeficient mice. This leads to rapid engraftment of mature human T cells but often results in a lethal xenogeneic graft-versus-host disease (GVHD) within weeks, limiting the experimental window.
- Hu-SRC-SCID: Model is established by engrafting human CD34+ haematopoietic stem cells (HSCs) sourced from bone marrow, cord blood, or fetal liver. This method results in a more complete and long-lasting reconstitution of a human immune system, including B cells, T cells, myeloid cells, and antigen-presenting cells.
- BLT (Bone Marrow, Liver, Thymus) Model: This is considered the gold standard for complexity. Human fetal liver and thymus tissue are transplanted under the mouse renal capsule, accompanied by an intravenous injection of autologous HSCs. BLT mice develop a robust, HLA-restricted human immune system with improved T-cell functionality, though they eventually develop GVHD, limiting the experimental timeframe.
Comparative Analysis of Humanized Mouse Models
The choice of humanization strategy significantly impacts the model's capabilities, strengths, and limitations. The table below provides a structured comparison to guide researchers in selecting the most appropriate system for their specific research questions, particularly in the context of infectious disease research.
Table 2: Comparative Analysis of Major Humanized Mouse Model Platforms
| Model Characteristic | Hu-PBL-SCID | Hu-SRC-SCID | BLT (Bone Marrow, Liver, Thymus) |
|---|---|---|---|
| Engraftment Method | Injection of human peripheral blood leukocytes (PBL) | Injection of human CD34+ hematopoietic stem cells | Co-transplantation of human fetal liver & thymus tissue + IV injection of CD34+ cells |
| Key Immune Components | Primarily mature human T cells | Multilineage human immune cells (B, T, myeloid, APC) | Complete, HLA-restricted human immune system with improved T-cell education |
| Primary Applications | Short-term T-cell studies, HIV infection models | Study of human hematopoiesis, immune response to pathogens | Studies requiring authentic human T-cell responses, mucosal immunity (e.g., HIV transmission) |
| Experimental Window | Short (weeks) due to lethal GVHD | Long-term (months) | Long-term, but eventually develops GVHD (~25-30 weeks) |
| Major Strength | Rapid reconstitution of functional human T cells | Multilineage reconstitution without HLA restriction | Most complete human immune system with HLA restriction; supports mucosal infection studies |
| Major Limitation | Limited lifespan; lacks broader immune reconstitution | T cells may not be fully functional due to lack of human thymic education | Technically complex; cost-intensive; develops GVHD |
The Scientist's Toolkit: Essential Research Reagents and Materials
The successful implementation of these advanced animal models requires specific biological reagents and technical instruments. The following table details the core components necessary for establishing and utilizing ultrasonic atomization and humanized mouse systems.
Table 3: Essential Research Reagents and Materials for Advanced Animal Models
| Item | Function & Application | Specific Examples / Specifications |
|---|---|---|
| Immunodeficient Mouse Strains | Serves as the in vivo recipient for human cell/tissue engraftment. | NOD.Cg-PrkdcscidIl-2rγtm1Wjl (NSG), Rag2nullIL-2rγnull, Fahnull/Rag2null/IL-2rγnull (FRG) [64]. |
| Human Biological Samples | Source for creating the human immune system in mice. | Peripheral Blood Leukocytes (PBLs), CD34+ Haematopoietic Stem Cells (from cord blood, bone marrow, fetal liver), fetal liver and thymus tissue [64]. |
| Ultrasonic Nebulizer | Generates a fine, inhalable aerosol of a bacterial suspension for establishing pulmonary infection models. | 402A1 type Ultrasonic Nebulizer; used to create a mouse lung infection model for MDR A. baumannii [63]. |
| Low-Frequency Ultrasound Device | Used as an adjuvant therapy to enhance penetration of antimicrobials (e.g., photosensitizers, antibiotics) into biofilms. | LIPUSTIM Sonodynamic Therapy solutions; 1 MHz frequency, 500 mW/cm² intensity [65]. |
| Cationic Photosensitizer (P3) | A novel antimicrobial photosensitizer used in antimicrobial photodynamic therapy (aPDT) studies, often in combination with ultrasound. | Cationic benzylidene cyclopentanone; exhibits high selectivity for bacterial over mammalian cells [65]. |
Discussion: Integrated Application and Correlation with Clinical Outcomes
The strategic integration of both ultrasonic atomization and humanized mouse models provides a powerful, multi-faceted approach to preclinical anti-infective development. Ultrasonic atomization models offer high reproducibility for screening therapeutics against respiratory infections, especially those involving biofilms. The demonstrated synergy between minocycline and other antibiotics in this model [63] highlights its predictive value for identifying effective combination regimens for multi-drug resistant infections. Furthermore, the adjunctive use of ultrasound to enhance drug delivery, as shown in the synergistic effect of ultrasound with aPDT against MRSA biofilms [65], represents a promising avenue for overcoming biofilm-mediated treatment failure.
Humanized mouse models address the critical limitation of species-specificity. They have become a preclinical gold standard for investigating human-tropic pathogens like HIV and for evaluating immunotherapies [64] [66]. The ability of the BLT model, for instance, to support rectal and vaginal transmission of HIV provides an unparalleled platform for studying prevention strategies [64]. The growing market for these models, with mice accounting for a 65% share due to their genetic tractability and physiological relevance, underscores their entrenched value in biomedical research [67].
Ultimately, the correlation between in vitro and in vivo efficacy is greatly enhanced by employing disease models that more accurately reflect human physiology and pathology. While traditional in vitro systems often fail to predict clinical success due to their oversimplification [41], the advanced animal models detailed in this guide provide a critical intermediary step. They enable researchers to dissect complex host-pathogen-therapy interactions in a controlled yet physiologically relevant environment, thereby increasing the likelihood of clinical success for novel anti-infective agents and therapeutic strategies.
Validating Correlations and Comparative Analysis Across Anti-Infective Classes
In the field of anti-infective drug development, the relationship between in vitro activity and in vivo efficacy is foundational. While in vitro susceptibility testing provides a controlled, quantitative measure of a compound's activity against pathogens, the true therapeutic potential is only revealed within the complex biological environment of a living organism. Validation frameworks serve as the critical bridge connecting these two domains, ensuring that laboratory measurements reliably predict clinical outcomes. The pressing global antimicrobial resistance (AMR) crisis, affecting 2.8 million Americans annually, underscores the urgent need for accurate, predictive susceptibility testing methods that can keep pace with evolving pathogens [68]. This guide examines the key elements of these validation frameworks, comparing their application across in vitro and in vivo settings to support robust antimicrobial development.
Foundational Concepts: In Vitro and In Vivo Testing Paradigms
Defining the Testing Environments
In vitro studies (Latin for "in glass") are conducted outside living organisms using isolated cells, tissues, or biological molecules in controlled laboratory settings. These experiments allow researchers to investigate specific aspects of biological systems with precision and reproducibility, removing confounding factors present in whole organisms [69]. In antimicrobial development, this primarily involves susceptibility testing against pathogen cultures.
In vivo studies ("within the living") are performed within whole living organisms, such as animals or humans. These investigations observe biological processes in their natural, holistic context, providing high physiological relevance by capturing interactions between different organ systems and long-term responses to interventions [69]. In antimicrobial research, this typically involves animal infection models before progressing to human clinical trials.
Complementary Roles in Drug Development
Both approaches serve complementary, sequential roles in the drug development pipeline:
- In vitro studies enable high-throughput screening of potential drug candidates, mechanism of action studies, and initial potency assessments [69] [70].
- In vivo studies evaluate safety, toxicity, and efficacy in complex biological systems, accounting for pharmacokinetic/pharmacodynamic (PK/PD) variables like absorption, distribution, metabolism, and excretion [70].
The controlled environment of in vitro testing provides efficiency and reproducibility, while in vivo testing offers biological complexity but with greater resource requirements and ethical considerations [70]. Success in preclinical stages does not necessarily translate to clinical outcomes, highlighting the need for robust validation frameworks throughout the development process [70].
Validation Frameworks: The V3 Framework for Preclinical Research
Adaptation of Clinical Validation Standards
The validation of antimicrobial testing methods follows a structured framework adapted from clinical diagnostics. The Digital Medicine Society's (DiMe) V3 Framework—encompassing Verification, Analytical Validation, and Clinical Validation—provides a comprehensive approach to building evidence supporting the reliability and relevance of quantitative measures [71]. This framework has been adapted for preclinical research as the "In Vivo V3 Framework" to address the unique requirements and variability of animal models [71].
Table 1: The V3 Validation Framework for Preclinical Antimicrobial Research
| Component | Definition | Preclinical Application |
|---|---|---|
| Verification | Ensures digital technologies accurately capture and store raw data | Confirming sensors and instruments properly record susceptibility data in variable laboratory/animal environments |
| Analytical Validation | Assesses precision and accuracy of algorithms transforming raw data into biological metrics | Validating that algorithms correctly interpret zone of inhibition, MIC values, or bacterial burden reduction |
| Clinical Validation | Confirms measures accurately reflect biological/functional states in relevant animal models | Demonstrating that efficacy measures correlate with pathogen clearance or animal survival in infection models |
Regulatory Context and Recent Developments
Validation occurs within a strict regulatory landscape. In early 2025, the U.S. Food and Drug Administration (FDA) recognized many breakpoints published by the Clinical Laboratory Standards Institute (CLSI), including for microorganisms representing an unmet need [68]. This unprecedented step provides a pragmatic solution for antimicrobial susceptibility testing (AST) by clinical laboratories and marks a significant advancement for combating AMR [68].
Similarly, the Indian Council of Medical Research (ICMR) published 2025 guidance establishing a comprehensive framework for validating rapid diagnostics for pathogen identification and AST, aligning with Medical Device Rules, 2017, and international standards including ISO 20916:2019 and ISO 15189:2022 [72].
Quantitative Correlations Between In Vitro and In Vivo Activity
Experimental Models for Correlation Studies
The relationship between in vitro potency and in vivo efficacy has been systematically evaluated using animal infection models. A seminal murine thigh infection model study examined this correlation against 15 gram-negative bacilli from five different species using four antimicrobial agents with different mechanisms of action: tobramycin, pefloxacin, ceftazidime, and imipenem [73].
Researchers defined three key parameters of in vivo activity:
- Maximal attainable antimicrobial effect (Emax): Reduction in log₁₀ CFU per thigh compared with untreated controls at 24 hours
- Potency dose (P50): Total dose required to reach 50% of maximal effect
- Static dose: Total dose required to achieve a bacteriostatic effect
Table 2: Correlation Between In Vitro and In Vivo Parameters for Antimicrobial Agents [73]
| Antimicrobial Agent | Emax (Log₁₀ CFU Reduction) | Static Dose/MIC Ratio | Correlation Between In Vitro MIC and In Vivo Efficacy |
|---|---|---|---|
| Pefloxacin | Greatest reduction (P < 0.05) | Intermediate | Significant correlation for most strains |
| Tobramycin | Intermediate | Lowest (P < 0.002) | Significant correlation for most strains |
| Ceftazidime | Intermediate | Intermediate | Significant correlation for most strains |
| Imipenem | Intermediate | Higher against Enterobacteriaceae | No significant correlation (P > 0.50); greater potency against P. aeruginosa |
Factors Explaining Discrepancies
The study revealed that while in vitro susceptibility tests generally correlated well with in vivo activity in this animal model, important exceptions existed. Imipenem showed no significant correlation between its in vitro MIC and in vivo efficacy parameters, primarily due to its greater potency against Pseudomonas aeruginosa strains compared to Enterobacteriaceae (P < 0.01) [73]. This increased potency against P. aeruginosa was attributed to a longer post-antibiotic effect (P = 0.02), highlighting how pharmacodynamic properties can significantly influence the in vitro-in vivo relationship [73].
Variations in potency among the four antimicrobial agents were explained by differences in pharmacokinetics or pharmacodynamic activity, emphasizing that in vitro potency alone cannot fully predict in vivo efficacy without considering these additional factors [73].
Methodologies and Experimental Protocols
Murine Thigh Infection Model Protocol
The murine thigh infection model provides a standardized methodology for evaluating in vivo correlation with in vitro results [73]:
- Animal Preparation: Immunocompromised mice are prepared to ensure consistent infection establishment.
- Infection Induction: Both thighs are inoculated with a standardized inoculum (approximately 10⁶ CFU) of the test organism.
- Antimicrobial Dosing: Treatment begins 2 hours post-infection with complete dose-response curves determined for each antimicrobial against each strain.
- Sample Collection: Thighs are harvested 24 hours after infection initiation.
- Bacterial Burden Quantification: Homogenized thigh samples are plated for quantitative culture, with results expressed as log₁₀ CFU per thigh.
- Dose-Response Analysis: The dose-response relationship is analyzed using non-linear regression to determine Emax, P50, and static dose parameters.
In Vitro Susceptibility Testing Methods
Standardized in vitro methods provide the foundation for correlation with in vivo activity:
- Broth Microdilution: The CLSI-reference method performed according to M07 guidelines [68].
- Agar-Based Methods: Disk diffusion and gradient diffusion methods.
- Automated Systems: FDA-cleared automated susceptibility testing devices.
- Quality Control: Regular testing of control strains to ensure accuracy and precision.
Recent regulatory changes now recognize the CLSI broth microdilution method as described in M07 as a reference method used for device clearance [68].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for Antimicrobial Validation Studies
| Reagent/Material | Function in Validation | Application Context |
|---|---|---|
| Cation-adjusted Mueller-Hinton broth | Standardized medium for broth microdilution | In vitro susceptibility testing |
| Reference bacterial strains | Quality control for susceptibility testing | Both in vitro and in vivo studies |
| Animal models (e.g., murine) | Provide complex host environment for efficacy studies | In vivo infection models |
| Cell culture media | Support growth of bacterial pathogens | In vitro susceptibility testing |
| Antimicrobial standards | Reference compounds for potency comparison | Both in vitro and in vivo studies |
| Protein binding reagents | Simulate protein binding effects in human physiology | In vitro PK/PD modeling |
| Histology reagents | Tissue processing and staining for pathology assessment | In vivo studies |
| Molecular biology kits | Genomic analysis of pathogen response | Both in vitro and in vivo studies |
Analysis of Discrepancies and Resolution Strategies
Despite generally good correlation, several factors can contribute to discrepancies between in vitro activity and in vivo response:
- Post-antibiotic effect: Prolonged antimicrobial activity after drug removal, as observed with imipenem against P. aeruginosa, enhances in vivo efficacy beyond what in vitro MIC values predict [73].
- Protein binding: Serum protein binding reduces freely available drug concentrations in vivo.
- Tissue penetration: Differential drug distribution to infection sites affects local concentrations.
- Host immune system interactions: The immune system works synergistically with antimicrobial activity in vivo.
- Inoculum effect: Higher bacterial densities in vivo may reduce apparent drug efficacy.
Broader Evidence Across Drug Classes
A comprehensive survey of 164 marketed small-molecule drugs examined the general relationship between clinical unbound concentrations and in vitro potency [74]. The analysis revealed that approximately 70% of compounds had therapeutic unbound plasma exposure lower than in vitro potency, with a median ratio of exposure in relation to in vitro potency of 0.32 [74]. The range of ratios was substantial (0.007 to 8.7), highlighting the variability in predicting therapeutic exposure from in vitro data alone [74].
This analysis identified differences in the in vivo-to-in vitro potency ratio between therapeutic indications, mode of action, target type, and whether drugs had active metabolites [74]. These findings emphasize that generic predictions of in vivo efficacious concentrations based solely on in vitro potency may be highly variable and lack biological significance without considering these additional factors [74].
Integrated Workflow for Antimicrobial Validation
The following workflow diagram illustrates the integrated validation process for antimicrobial testing methods, incorporating both in vitro and in vivo components:
Diagram 1: Integrated validation workflow for antimicrobial testing methods
Validation frameworks for in vitro and in vivo antimicrobial testing serve as essential bridges between laboratory measurements and clinical efficacy. While significant correlations exist between in vitro susceptibility parameters and in vivo outcomes in animal models, the relationship is influenced by multiple factors including pharmacokinetic properties, pharmacodynamic characteristics, and specific pathogen-drug interactions. The adaptation of structured validation frameworks like the V3 approach, coupled with standardized experimental models and consideration of recent regulatory developments, provides a robust foundation for advancing antimicrobial development. As the AMR crisis continues to evolve, these validation frameworks will play an increasingly critical role in ensuring that new anti-infective therapies demonstrate predictable transitions from laboratory measurements to clinical efficacy, ultimately supporting more effective patient care and antimicrobial stewardship.
The evaluation of anti-infective efficacy presents a significant challenge in pharmaceutical development, particularly concerning microorganisms in dormant physiological states or complex communities. Standard in vitro susceptibility tests, designed against planktonic, rapidly dividing bacteria, frequently fail to predict therapeutic outcomes for chronic infections [75] [76]. This case study examines the critical correlation—and frequent disconnect—between in vitro and in vivo efficacy of antimicrobial agents against stationary-phase and adherent microbial populations, with a focus on its implications for drug development.
The core of the problem lies in the inherent limitations of planktonic models. Biofilms, structured communities of microorganisms encased in a self-produced polymeric matrix, are estimated to be involved in over 80% of chronic and recurrent human infections [76]. Cells within a biofilm demonstrate major physiological changes compared to their planktonic counterparts and can be 100 to 1000 times less susceptible to antimicrobial agents [44] [77]. This discrepancy necessitates more predictive testing models that account for the biofilm mode of growth.
Key Concepts and Definitions
Understanding the following concepts is essential for interpreting efficacy correlations:
- Planktonic Cells: Free-living, single microbial cells suspended in a liquid medium. This is the phenotype targeted by conventional antibiotic susceptibility tests (e.g., MIC, MBC determinations) [75].
- Stationary-Phase Cells: Microbes that have entered a state of slow or non-growth due to nutrient depletion or other environmental stresses. This state often leads to increased phenotypic tolerance to antimicrobials [78] [75].
- Adherent Cells/Biofilms: Microbial communities attached to a surface and embedded in an extracellular polymeric substance (EPS) matrix. This lifestyle confers profound resistance through multiple mechanisms [76] [77].
- Germicidal Effect (GE): A quantitative measure of disinfectant or antimicrobial efficacy, often calculated as the log reduction in viable counts after treatment:
GE = log(Nc) - log(Nd), where Nc is the number of untreated bacteria and Nd is the number after treatment [79].
Foundational In Vitro/In Vivo Correlation Study
A seminal 1990 study by et al. provided a rigorous investigation into the correlation between standard in vitro tests and in vivo efficacy for device-related infections [78] [75].
Experimental Methodology
- In Vivo Model: The guinea pig tissue-cage model was used. Sterile perforated tubes filled with sinter-glass beads were implanted subcutaneously in guinea pigs. After healing, the cages were infected with a methicillin-resistant Staphylococcus epidermidis strain, and antibiotic therapy was initiated 16 hours post-inoculation [75].
- In Vitro Models: Several tests were performed:
- Pharmacokinetic Matching: The in vitro model was designed to simulate the exact kinetic concentrations of antibiotics measured in the tissue-cage fluid of the animal model, allowing for a direct comparison [75].
Key Findings and Correlation Analysis
The study yielded critical insights into which in vitro tests best predicted in vivo success.
Table 1: Correlation of In Vitro Efficacy with In Vivo Outcomes in a Foreign Body Infection Model [75]
| Antibiotic Tested | Efficacy on Stationary & Adherent Cells (In Vitro) | Eradication in Animal Model (In Vivo) | Correlation |
|---|---|---|---|
| Rifampin | Highly efficient | 12/12 infections cured | Strong |
| Ciprofloxacin | Low efficacy | Failure to eradicate | Strong |
| Amikacin | Not effective | 0/24 beads sterilized | Strong |
| Levofloxacin | Variable efficacy (strain-dependent) | 1/24 (RP62A) vs 8/24 (M7) beads sterilized | Moderate |
| Teicoplanin | Not effective | 0/24 beads sterilized | Strong |
The data demonstrated that standard MICs for planktonic cells were not predictive of therapeutic success in this biofilm-related infection. In contrast, drug efficacy on stationary-phase and adherent microorganisms successfully predicted the outcome of device-related infections [78] [75]. For instance, rifampin, which cured all 12 infections in vivo, was also the most effective drug in all in vitro tests against non-growing and adherent cells.
Extended Experimental Evidence and Data
Subsequent research has consistently reinforced these findings across different pathogens and antimicrobial classes.
Efficacy of Disinfectants on Planktonic vs. Biofilm Cells
A 2023 study compared the efficacy of a peracetic acid-based disinfectant (P) and a benzalkonium chloride-based disinfectant (D) against planktonic and biofilm populations of Staphylococcus aureus and Escherichia coli [79].
Experimental Protocol:
- Planktonic Testing: A quantitative suspension test was used based on DGHM guidelines. Bacteria were exposed to disinfectants (0.1%, 0.3%, 0.5%) for 5 and 10 minutes, and viable counts were determined on TSA with neutralizers [79].
- Biofilm Formation: Biofilms were grown on polystyrene microtiter plates for 48 hours at 25°C, and production was confirmed with a crystal violet test [79].
- Biofilm Efficacy: The germicidal effect of the same disinfectant concentrations was determined on 48-hour biofilms [79].
Table 2: Comparative Efficacy of Disinfectants on Planktonic vs. Biofilm Cells [79]
| Microorganism | Disinfectant | Concentration for 100% GE on Planktonic Cells | GE on 48h Biofilms | Concentration for Complete Biofilm Destruction |
|---|---|---|---|---|
| S. aureus | Peracetic (P) | 0.1% for 5 min | Significantly weaker | 2% for 5 min |
| E. aureus | Benzalk. (D) | 0.1% for 5 min | Significantly weaker | 2% for 5 min |
| E. coli | Peracetic (P) | 0.1% for 5 min | Significantly weaker | 2% for 5 min |
| E. coli | Benzalk. (D) | 0.1% for 5 min | Significantly weaker | 2% for 5 min |
This study highlighted that both disinfectants showed a significantly weaker germicidal effect on biofilms compared to planktonic cells, requiring a 20-fold higher concentration (2% vs. 0.1%) to achieve complete destruction of viable biofilm cells [79].
Novel Anti-Biofilm Strategies
The challenge of biofilm resistance has spurred the development of innovative therapeutic strategies.
- Guanidinium-Linked Neomycin Lipidation (2025): A novel conjugate was designed to overcome neomycin's limited efficacy. This construct integrates a neomycin core, a hydrophobic lipid chain, and a cationic guanidinium moiety. The result is a compound with significantly improved antibacterial activity and the ability to effectively disrupt biofilm formation in vitro and in vivo against both Gram-positive and Gram-negative pathogens [80].
- Thymol against Cross-Kingdom Biofilms (2021): Thymol, a plant-derived monoterpene, demonstrated potent anti-biofilm activity against dual-species biofilms of Candida albicans and Streptococcus mutans, a combination relevant to early childhood caries. Thymol at 300 μg/mL arrested growth and proliferation and, notably, showed rapid killing efficacy within 2 minutes in a time-kill assay. It also reduced key virulence factors like hyphal transition in Candida and acid production in Streptococci [81].
Mechanisms Underlying the Efficacy Gap
The profound tolerance of biofilms and stationary-phase cells to antimicrobials is not due to a single mechanism but a combination of collaborative factors.
Diagram: Collaborative Mechanisms of Biofilm-Associated Antimicrobial Tolerance
The mechanisms can be categorized as follows [76] [77]:
- Physical Barrier and Sorption: The extracellular polymeric substance (EPS) matrix, composed of polysaccharides, proteins, and extracellular DNA (eDNA), can act as a barrier, slowing antibiotic diffusion. Positively charged antibiotics (e.g., aminoglycosides) can bind to negatively charged polymers like eDNA, preventing them from reaching cellular targets [77].
- Altered Microenvironment: Metabolic activity within the biofilm creates chemical gradients (e.g., of nutrients and oxygen), leading to zones of slow growth or metabolic dormancy. Since many antibiotics require active cell growth, these dormant cells are protected [76]. Fermentation can also create an acidic environment that reduces the activity of certain antibiotic classes [76].
- Physiological Heterogeneity and Persistence: Biofilms contain a heterogeneous mix of cells at different metabolic states. A subpopulation of "persister" cells enters a deep dormant state, exhibiting extreme tolerance to antimicrobials. These cells are not mutants but phenotypic variants capable of repopulating the biofilm after treatment ceases [76] [44].
- Enhanced Horizontal Gene Transfer (HGT): The close proximity of cells within the biofilm matrix facilitates the efficient exchange of plasmids and other mobile genetic elements, accelerating the spread of conventional antibiotic resistance genes (e.g., those encoding efflux pumps or drug-modifying enzymes) [77].
The Scientist's Toolkit: Essential Research Reagents and Models
To effectively study biofilm efficacy and in vitro-in vivo correlation, researchers rely on a suite of specialized tools and models.
Table 3: Key Reagents and Models for Anti-Biofilm Efficacy Research
| Item Category | Specific Example(s) | Function & Application in Research |
|---|---|---|
| In Vitro Biofilm Models | Microtiter Plate Crystal Violet Assay [79], MBEC (Minimum Biofilm Eradication Concentration) Assay [44], Robbins Device [44] | High-throughput screening of antimicrobial efficacy against pre-formed biofilms. |
| In Vivo Infection Models | Guinea Pig Tissue-Cage Model [75], Catheter-Associated Infection Models [44] | Mimics device-related biofilm infections in a living host to validate in vitro findings. |
| Specialized Culture Media | PBS-GCP [Phosphate-Buffered Saline with Glucose, Casamino Acids, Plasma] [75] | Supports limited bacterial growth to simulate slow-growing, stationary-phase conditions relevant to in vivo infections. |
| Biofilm Disruption Agents | Glycoside Hydrolases [77], Fibrinolytic Agents [77] | Enzymes that degrade specific components of the biofilm matrix (e.g., polysaccharides, fibrin), used to study dispersal and enhance antimicrobial penetration. |
| Neutralizing Agents | Lecithin (3 g/L) & Polysorbate 80 (30 g/L) [79] | Added to culture media to neutralize the residual effect of disinfectants or antimicrobials after contact time, allowing accurate counting of surviving cells. |
| Biosensor Strains | Chromobacterium violaceum CV026 [79] | A qualitative biosensor used in disc diffusion assays to determine if an antimicrobial has anti-quorum sensing activity, in addition to its direct antimicrobial effect. |
The correlation between in vitro efficacy and in vivo outcomes for anti-infective agents is profoundly influenced by the microbial growth phenotype. This case study underscores that standard planktonic susceptibility tests are inadequate for predicting the efficacy of treatments against biofilm-associated and chronic infections [78] [75] [76].
To improve the predictive power of preclinical research, the field must adopt more sophisticated models. Key recommendations include:
- Utilizing Biofilm-Specific In Vitro Models: Methods like the MBEC assay and other biofilm reactors that generate standardized, mature biofilms for screening are essential [44].
- Incorporating Stationary-Phase Killing Assays: Evaluating efficacy against slow-growing or non-growing cells provides critical data that better correlates with in vivo efficacy for many chronic infections [78] [75].
- Bridging with Pharmacokinetically-Relevant In Vivo Models: Animal models that replicate key aspects of human infection, such as foreign body colonization, and whose pharmacokinetics can be mirrored in vitro, provide the strongest bridge for translational research [75] [44].
Future success in developing anti-infectives against resilient biofilm infections will depend on a commitment to these more complex, but more predictive, efficacy correlations. The scientific toolkit is available; its consistent and rigorous application is the path forward.
The translation of drug efficacy from laboratory models to clinical success is a pivotal challenge in anti-infective development. In vitro-in vivo correlation (IVIVC) plays a crucial role in this process, yet its application and predictive power differ dramatically between viral and bacterial infections. For influenza antivirals, strong IVIVC has significantly accelerated and optimized drug development, leading to a robust pipeline of effective treatments. In contrast, bacterial infection models face substantial biological and methodological hurdles that often result in poor clinical translation. This comparative analysis examines the underlying factors contributing to this disparity, providing researchers with experimental data, methodological insights, and visual tools to navigate these distinct developmental landscapes.
IVIVC Success in Influenza Antiviral Development
Established Models and Strong Correlations
The development of influenza antivirals has benefited from predictive laboratory models that successfully mirror human infection dynamics. Cell-based assays and animal models have demonstrated remarkable accuracy in predicting human pharmacokinetic and pharmacodynamic parameters for several drug classes.
Table 1: Successful IVIVC in Approved Influenza Antivirals
| Drug (Class) | In Vitro System | Key Predictive Parameters | In Vivo Validation | Correlation Strength |
|---|---|---|---|---|
| Peramivir (NAI) | A549 cell uptake/transport | OCTN2 transporter substrate identification | Rat lung tissue distribution [82] | Strong (R² >0.8) |
| Peramivir (NAI) | Caco-2/MDCK permeability | Low permeability, no efflux transport | Restricted lung penetration after inhalation [82] | Strong |
| Favipiravir (RdRp) | Dissolution testing (pH 6.8) | AUC0-t vs. % dissolved | Level C IVIVC in Egyptian volunteers [83] | Strong |
A compelling example comes from peramivir inhalation studies, where in vitro models accurately predicted in vivo lung distribution patterns. Research demonstrated that peramivir exhibited low permeability across diverse cell systems with no participation of efflux transporters, correctly forecasting its predominant localization within alveolar epithelial lining fluid and minimal systemic dissemination after airway inhalation in rats [82]. This precise IVIVC enabled researchers to optimize inhalation formulations that maximize target site exposure while reducing systemic exposure—a critical advantage for respiratory antivirals.
Methodological Protocols for Influenza Antiviral Assessment
Standardized experimental workflows contribute significantly to reliable IVIVC in influenza drug development:
Cellular Uptake and Transport Studies
- Cell Models: A549 (human lung adenocarcinoma), Caco-2, MDCK
- Methodology: Cells cultured on permeable supports, drug applied to donor compartment, samples collected from receiver compartment over time
- Analysis: LC-MS/MS quantification, apparent permeability calculation, transporter inhibition studies
- Duration: 4-6 hours sampling, with full study spanning 2-3 days [82]
In Vivo Pharmacokinetic Studies
- Animal Model: Sprague-Dawley rats (250-300g)
- Dosing: Intravenous (3-6 mg/kg) vs. intratracheal inhalation (0.3-6 mg/kg)
- Sample Collection: Serial blood sampling via catheter, bronchoalveolar lavage at endpoint, tissue collection (lung, liver, kidney)
- Analysis: Non-compartmental PK analysis, tissue distribution quantification [82]
Dissolution-IVIVC Correlation
- Media: Phosphate buffer (pH 6.8), simulated gastric/intestinal fluids
- Apparatus: USP dissolution apparatus II (paddle)
- Correlation: Level C IVIVC establishing point-to-point relationship between dissolution efficiency and AUC0-t [83]
Figure 1: Successful IVIVC workflow for influenza antivirals demonstrating strong correlation points from cellular assays to clinical translation.
IVIVC Challenges in Bacterial Infection Models
Fundamental Limitations in Bacterial Disease Modeling
Unlike influenza models, bacterial infection research faces substantial obstacles in achieving predictive IVIVC. Traditional in vitro systems fail to replicate the complex host-pathogen interactions occurring in human infections, leading to frequent clinical trial failures.
Table 2: Key Challenges in Bacterial Infection IVIVC
| Challenge Category | Specific Limitations | Impact on IVIVC | Examples |
|---|---|---|---|
| Model Complexity | Lack of host immune components | Poor prediction of antibiotic efficacy in vivo | Prontosil effective in mice but not in vitro [41] |
| Biofilm Microenvironment | Absence of EPS matrix in traditional models | Underestimation of antibiotic resistance | 10-1000x higher antibiotic tolerance in biofilms [41] |
| Species Differences | Murine vs. human immune responses | Misleading efficacy and safety data | Innate immune system organization differences [41] |
| PK/PD Discrepancies | Differing drug penetration and clearance | Inaccurate human dosing predictions | Varied tissue distribution across species [41] |
The biofilm complication represents a particularly significant challenge. Bacteria in biofilms can tolerate 10-1000 times higher antibiotic concentrations than their planktonic counterparts, but most conventional in vitro models fail to incorporate this critical aspect of human infections [41]. This discrepancy explains why many compounds showing excellent activity in standard microtiter plate assays demonstrate poor efficacy in clinical settings where biofilms dominate chronic and device-associated infections.
Methodological Gaps in Bacterial Infection Protocols
Current limitations in bacterial infection modeling methodologies include:
Traditional Susceptibility Testing Flaws
- Platform: Microtiter plate assays
- Missing Elements: Host proteins, fluid flow, biomechanical cues, intercellular interactions
- Outcome: Overestimation of antibiotic efficacy against planktonic bacteria only
- Duration: 24-48 hours, insufficient for biofilm formation studies [41]
Inadequate Biofilm Models
- Standard Approach: Static biofilm assays
- Limitations: Poor representation of in vivo biofilm architecture and matrix composition
- Missing Factors: Immune cell interactions, nutrient gradients, EPS component variations
- Solution Need: Advanced flow cell systems, organ-on-a-chip technologies [41]
Animal Model Discrepancies
- Immune Differences: Varied organization of murine vs. human immune systems
- Dosing Challenges: Differing pharmacokinetic profiles across species
- Infection Introduction: Artificial bacterial inoculation methods not mimicking human acquisition [41]
Comparative Analysis: Key Divergence Points
Biological and Methodological Distinctions
The disparity in IVIVC success between influenza antivirals and antibacterial drugs stems from fundamental differences in disease biology and model systems:
Figure 2: Key divergence points explaining IVIVC success in influenza models versus challenges in bacterial infection models.
Quantitative Disparities in Model Predictive Value
Table 3: Direct Comparison of IVIVC Performance Metrics
| Performance Indicator | Influenza Antiviral Models | Bacterial Infection Models |
|---|---|---|
| Model Accuracy | >80% prediction of human PK parameters [82] | 20-30% clinical trial success rate [41] |
| Translatable Efficacy | Strong dose-response correlation (R²=0.75-0.95) | Frequent efficacy overestimation (10-1000x) [41] |
| Resistance Prediction | Accurate NAI resistance profiling in cell culture | Poor prediction of biofilm-mediated resistance |
| Tissue Distribution | IVIVC successfully guides formulation (e.g., inhaled peramivir) [82] | Limited penetration prediction in abscesses/biofilms |
| Time to Clinical Use | Accelerated development (5-7 years) | Prolonged development (10-15 years) with high failure |
The data reveal that influenza antiviral development benefits from direct viral targeting and well-conserved mechanisms across model systems, whereas antibacterial development must account for complex bacterial communities (biofilms), diverse resistance mechanisms, and intricate host-pathogen interactions that are poorly replicated in standard models.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Key Materials for Anti-Infective IVIVC Research
Table 4: Essential Research Reagents for IVIVC Studies
| Reagent/Cell Line | Application | Function in IVIVC | Evidence Source |
|---|---|---|---|
| A549 Cells | Influenza antiviral uptake | Human lung epithelial model for drug transport | Peramivir uptake studies [82] |
| Caco-2/MDCK Cells | Permeability assessment | Intestinal/epithelial barrier function prediction | Transmembrane transport studies [82] |
| OCTN2 Transporter | Drug transport mechanisms | Identifies substrate-specific uptake pathways | Peramivir transporter identification [82] |
| 16S rRNA Sequencing | Microbiome analysis | Respiratory microbiome dysbiosis assessment | Influenza vs. bacterial signature identification [84] |
| IFI27 Biomarker | Host response discrimination | Differentiates viral vs. bacterial infection | 88% diagnostic accuracy in respiratory illness [85] |
| Biofilm Flow Cells | Bacterial biofilm models | Incorporates fluid dynamics into biofilm studies | Advanced infection model development [41] |
Emerging Solutions for Bacterial IVIVC Challenges
Promising approaches to address bacterial IVIVC limitations include:
Advanced Biofilm Models
- Technology: Organ-on-a-chip with fluid flow
- Advantage: Incorporates mechanical cues, immune components, and relevant growth matrices
- Application: Chronic infection modeling, medical device-associated infections [41]
Host-Directed Therapeutics
- Approach: Monoclonal antibodies, immunomodulators, virulence disruptors
- IVIVC Advantage: Less susceptible to conventional resistance mechanisms
- Challenge: Complex clinical trial design requirements [86]
Microbiome-Based Diagnostics
- Technology: Respiratory microbiome signature analysis
- Application: Differentiates influenza from bacterial infections via signature organisms
- Accuracy: Specific bacterial phyla identification (Firmicutes, Actinobacteria) [84]
This comparative analysis demonstrates that the established success of IVIVC in influenza antiviral development stems from predictive model systems that accurately replicate key aspects of human infection and drug response. In contrast, bacterial infection modeling faces fundamental challenges related to biofilm complexity, inadequate host factor incorporation, and species-specific immune responses that severely limit translational predictive value. Researchers pursuing antibacterial development must prioritize advanced model systems that better replicate human host environments, including biofilm-relevant conditions, immune components, and species-specific pharmacokinetics. The integration of these sophisticated approaches represents the most promising path toward achieving the level of IVIVC success currently enjoyed in influenza antiviral development.
The escalating crisis of antimicrobial resistance has intensified the need for robust methodologies to evaluate the efficacy of new anti-infective agents and combinations. In this context, accurately benchmarking efficacy through standardized in vitro parameters is a critical step in the research and development pipeline. These in vitro metrics serve as the foundational predictors of in vivo success, guiding decisions about which therapeutic candidates advance to costly and complex clinical trials. The correlation between in vitro potency and in vivo immunogenicity or efficacy is a central tenet of pharmaceutical development, especially for novel modalities like mRNA vaccines and antimicrobial peptides [21].
Among the various available metrics, the Fractional Inhibitory Concentration Index (FICI) and log reduction values derived from time-kill studies have emerged as cornerstone parameters for validating antimicrobial activity and synergy. The FICI paradigm provides a standardized framework for quantifying drug interactions, enabling researchers to distinguish between synergistic, additive, indifferent, and antagonistic effects [87]. Meanwhile, log reduction measurements offer a dynamic, quantitative perspective on the rate and extent of antimicrobial killing, providing critical insights that static minimum inhibitory concentration (MIC) values cannot capture [27]. This guide objectively compares the application, interpretation, and validation of these pivotal parameters within the broader context of establishing reliable correlations between in vitro findings and in vivo outcomes in anti-infective research.
Core Parameters for Efficacy Benchmarking
Fractional Inhibitory Concentration Index (FICI)
The FICI is a quantitative measure used to characterize the interaction between two or more antimicrobial agents when used in combination. It is calculated based on the principle of Loewe additivity, which assumes that a drug interacts with itself [88]. The index provides researchers with a standardized value to classify the nature of drug interactions, which is particularly valuable for screening synergistic combinations against multidrug-resistant (MDR) pathogens or biofilm-associated infections where single drugs often fail [88].
Calculation and Interpretation: The FICI is calculated using the formula: FICI = (MIC of drug A in combination / MIC of drug A alone) + (MIC of drug B in combination / MIC of drug B alone). The resulting value is interpreted according to established thresholds:
- FICI ≤ 0.5: Synergism
- 0.5 < FICI ≤ 4: Additivity or Indifference
- FICI > 4: Antagonism
Table 1: FICI Interpretation Guidelines
| FICI Range | Interpretation | Clinical Implication |
|---|---|---|
| ≤ 0.5 | Synergism | Combination is significantly more effective than either agent alone. |
| > 0.5 to ≤ 4 | Additivity/Indifference | Combined effect is additive or no significant interaction. |
| > 4 | Antagonism | Combination is less effective than the single most active agent. |
Experimental Protocol (Checkerboard Assay): The standard method for determining FICI is the checkerboard assay, which involves the following steps [89]:
- Preparation: A two-dimensional array of serial dilutions of the two antimicrobial agents is prepared in a liquid medium or agar in a multi-well plate.
- Inoculation: Each well is inoculated with a standardized bacterial suspension (typically 5 × 10⁵ CFU/mL).
- Incubation: The plate is incubated under appropriate conditions for 16-20 hours.
- Analysis: The MIC of each drug alone and in combination is determined visually or spectrophotometrically as the lowest concentration that completely inhibits growth.
- Calculation: The FICI is calculated for each combination that inhibits growth, and the lowest FICI value is reported.
Application Example: A recent study investigating the synergy between colistin (COL) and α-terpineol (α-TP) against colistin-resistant gram-negative bacteria demonstrated pronounced synergism. The combination reduced the MIC of colistin by 4- to 2,048-fold, with FICI values ranging from 0.046875 to 0.5, successfully restoring susceptibility in all tested strains [89].
Log Reduction from Time-Kill Studies
While FICI provides a snapshot of potency, log reduction quantifies the bactericidal or fungicidal activity of an antimicrobial agent over time. This parameter, derived from time-kill studies, measures the rate and extent of microbial killing, offering a dynamic view of efficacy that is crucial for predicting in vivo outcomes, especially for concentration-dependent antibiotics [27].
Calculation and Interpretation: Log reduction is calculated by comparing the microbial count before and after exposure to an antimicrobial agent. The formula is: Log₁₀ Reduction = Log₁₀ (Initial Viable Count) - Log₁₀ (Viable Count at Time t). A 1-log reduction equals a 90% kill rate, a 2-log reduction equals 99%, a 3-log reduction equals 99.9%, and so on. This metric is directly related to the concept of the Minimum Bactericidal Concentration (MBC), which is defined as the lowest concentration of an antibiotic that achieves a ≥99.9% (or 3-log) reduction in the initial inoculum [27].
Experimental Protocol (Time-Kill Assay): The time-kill study methodology involves tracking the decline in viable microbes over time [27]:
- Inoculation: Tubes containing a growth medium are inoculated with a standardized microbial suspension (typically 10⁵-10⁶ CFU/mL).
- Antibiotic Exposure: The antimicrobial agent is added at predetermined concentrations (e.g., 1x, 2x, 4x MIC).
- Incubation and Sampling: The tubes are incubated, and samples are withdrawn at specific time intervals (e.g., 0, 2, 4, 6, 8, 12, 24 hours).
- Quantification: Each sample is serially diluted and plated onto agar plates to count the number of viable colonies (CFU/mL) after incubation.
- Analysis: The log₁₀ CFU/mL is plotted against time for each concentration, and the log reduction is calculated at each time point.
Application Example: In the study of colistin and α-terpineol, time-kill assays demonstrated that the combination achieved a ≥2 log₁₀ CFU/mL reduction against most bacterial strains within 6–12 hours of treatment, a significantly more rapid and extensive kill than either monotherapy [89].
Table 2: Comparison of Key Efficacy Parameters
| Parameter | FICI (from Checkerboard Assay) | Log Reduction (from Time-Kill Study) |
|---|---|---|
| Primary Purpose | Quantifies drug interaction (synergy, additivity, antagonism) | Quantifies the rate and extent of microbial killing (cidal activity) |
| Type of Measure | Static endpoint (after 16-20 hrs) | Dynamic, time-dependent profile |
| Key Output | Numerical index (FICI value) | Log₁₀ reduction in CFU/mL at specific time points |
| Strengths | Standardized, high-throughput synergy screening | Reveals kinetics of killing; can detect tolerance and regrowth |
| Limitations | Fixed concentration ratio may not reflect changing in vivo ratios; single time point. | Labor-intensive; does not simulate changing drug concentrations in vivo. |
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful execution of these efficacy benchmarks relies on specific reagents and tools. The following table details key materials and their functions in the described experimental protocols.
Table 3: Essential Research Reagents and Materials for Efficacy Testing
| Reagent / Material | Function in Experimental Protocol |
|---|---|
| Cation-Adjusted Mueller-Hinton Broth (CAMHB) | Standard medium for antibacterial susceptibility testing, providing consistent ion concentrations for reliable results. |
| 96-Well Microtiter Plates | Platform for performing high-throughput checkerboard assays and determining Minimum Inhibitory Concentrations (MICs). |
| Digital Colony Counter | Enables accurate and efficient counting of Colony Forming Units (CFUs) from time-kill assay plates for log reduction calculations. |
| Sterile Phosphate Buffered Saline (PBS) | Used for serial dilutions of bacterial suspensions and samples from time-kill studies prior to plating. |
| Reactive Oxygen Species (ROS) Detection Kits | Probe the mechanism of action of antimicrobials or adjuvants (e.g., menadione) by measuring oxidative stress in bacterial cells [88]. |
| Crystal Violet Stain | Standard dye used in biofilm assays to quantify total biofilm biomass, assessing both biofilm inhibition and eradication [89] [90]. |
Experimental Workflows and Correlation with In Vivo Outcomes
The following diagram visualizes the integrated experimental workflow for benchmarking efficacy, from initial in vitro screening to the critical step of correlating with in vivo models.
Navigating the Correlation Challenge: Establishing a predictable correlation between in vitro potency and in vivo efficacy remains a significant hurdle. While in vitro models are cost-effective, reproducible, and avoid ethical concerns of animal testing, they cannot fully replicate the complex physiology of a living host, including immune responses, tissue penetration, and pharmacokinetic (PK) variability [91]. For instance, an in vitro potency assay for an RSV antigen was found to be more stringent than the corresponding in vivo immunogenicity assay in mice, highlighting that the correlation is not always 1:1 [21].
Strategies for Enhanced Prediction: To improve predictive power, the field is moving toward more sophisticated models. These include:
- Advanced In Vitro Models: Utilizing hollow fiber infection models (HFIM) that simulate human PK profiles, or 3D cell cultures that better mimic tissue architecture, can bridge the gap between static in vitro assays and in vivo conditions [27] [91].
- Mechanistic Studies: Combining FICI and log reduction data with mechanistic insights (e.g., ROS production, membrane disruption) provides a more comprehensive understanding of agent action, aiding in vivo study design [89] [88].
- PK/PD Integration: Incorporating pharmacokinetic/pharmacodynamic (PK/PD) principles is pivotal. Parameters like the time that drug concentration remains above the MIC, or the ratio of peak concentration to MIC, are derived from in vitro data but are used to model and predict in vivo dosing regimens and efficacy [27].
The ultimate goal is a robust framework where in vitro benchmarks like FICI and log reduction, complemented by secondary and mechanistic assays, provide a reliable gateway for advancing the most promising anti-infective strategies toward clinical success.
Conclusion
The successful correlation of in vitro and in vivo anti-infective efficacy is not a singular achievement but a continuous process of model refinement. The key takeaway is that simplistic in vitro models are insufficient; predictive power is dramatically enhanced by incorporating physiological relevance, such as biofilm assays, and leveraging advanced mathematical PK/PD modeling. The future of IVIVC lies in the wider adoption of these sophisticated tools, including semi-mechanistic models that account for xenograft-specific parameters and exposure dynamics. Furthermore, as combination therapies and novel agents against multi-drug resistant pathogens evolve, robust IVIVC frameworks will be indispensable for prioritizing candidates, optimizing dosing regimens, and accelerating the development of effective new anti-infectives, ultimately bridging the translational gap between the laboratory and the clinic.
References
- 1. Comparison of the in vivo efficacy and resistance ... [https://pubmed.ncbi.nlm.nih.gov/40042366/]
- 2. From In Vitro Promise to In Vivo Reality: An Instructive ... [https://www.mdpi.com/1422-0067/25/18/9773]
- 3. Research progress on bacterial outer membrane vesicles ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1670307/full]
- 4. An in vitro study on the antimicrobial efficacy of a calcium ... [https://link.springer.com/article/10.1007/s00784-025-06524-w]
- 5. In vitro and In Vivo Antimicrobial Activity of Melia ... [https://pubmed.ncbi.nlm.nih.gov/41085779/]
- 6. Broad-spectrum antimicrobial activity and in vivo efficacy of ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1553693/full]
- 7. Developing In Vitro–In Vivo Correlation for Bicalutamide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12473495/]
- 8. Improving In Vitro–In Vivo Correlation (IVIVC) for Lipid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12567226/]
- 9. CMC Perspectives of In Vitro / In Vivo Correlation (IVIVC) ... [https://premier-research.com/perspectives/cmc-perspectives-of-in-vitro-in-vivo-correlation-ivivc-in-drug-development/]
- 10. Targeting Bacterial Biofilms on Medical Implants [https://pmc.ncbi.nlm.nih.gov/articles/PMC12382947/]
- 11. Discrepancies between in vitro activity of and in vivo ... [https://pubmed.ncbi.nlm.nih.gov/6423340/]
- 12. Biofilms: Understanding the structure and contribution ... [https://www.sciencedirect.com/science/article/pii/S2590097823000095]
- 13. Biofilm antimicrobial susceptibility through an experimental ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9579162/]
- 14. Is Increased Biofilm Formation Associated with Decreased ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12566419/]
- 15. An anaerobic in vitro flow model for studying interactions at ... [https://www.nature.com/articles/s41522-025-00800-z]
- 16. In Vitro Resistance-Predicting Studies and In ... [https://www.mdpi.com/1424-8247/17/8/1068]
- 17. Combating Bacterial Biofilms: Current and Emerging ... [https://www.mdpi.com/2079-6382/12/6/1005]
- 18. Model-based translation of results from in vitro to in vivo ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11638087/]
- 19. Importance of Relating Efficacy Measures to Unbound ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3623378/]
- 20. Issues in Pharmacokinetics and Pharmacodynamics of Anti ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC400530/]
- 21. Establishing correlation between in vitro potency and in ... [https://www.nature.com/articles/s41541-025-01181-2]
- 22. Application and development of Organ-on-a-Chip technology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12446035/]
- 23. Human organs-on-chips for disease modelling, drug ... [https://www.nature.com/articles/s41576-022-00466-9]
- 24. A guide to the organ-on-a-chip [https://www.nature.com/articles/s43586-022-00118-6]
- 25. State-of-the-art in high throughput organ-on-chip for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12149869/]
- 26. Expanding the Frontiers of Human Biology with Organ-on-a ... [https://emulatebio.com/expanding-the-frontiers-of-human-biology-with-organ-on-a-chip-technology-highlights-from-the-2025-mps-world-summit/]
- 27. Enhancing antibiotic therapy through comprehensive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11893874/]
- 28. Application of MIDD to accelerate the development of anti- ... [https://www.sciencedirect.com/science/article/abs/pii/S0169409X24002692]
- 29. Establishing in vitro-in vivo correlation for antibody drug ... [https://pubmed.ncbi.nlm.nih.gov/29423862/]
- 30. Translation of the efficacy of antibody–drug conjugates from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10339704/]
- 31. Semi-Mechanistic Model for the Antitumor Response of a ... [https://www.mdpi.com/2072-6694/13/20/5049]
- 32. A Review of Mathematical Models for Tumor Dynamics and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6813171/]
- 33. Pharmacokinetic/pharmacodynamic models for time ... [https://www.sciencedirect.com/science/article/pii/S0924857922001285]
- 34. Overall in vitro, in vivo, and in silico evaluation of Olea ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1567921/full]
- 35. Pharmacokinetic and Pharmacodynamic Principles of Anti ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5039113/]
- 36. In vitro-In vivo Correlation: Perspectives on Model Development [https://pmc.ncbi.nlm.nih.gov/articles/PMC3099258/]
- 37. Antibody–Drug Conjugates (ADCs): current and future ... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-025-01704-3]
- 38. Antibody-drug conjugates as multimodal therapies against ... [https://www.sciencedirect.com/science/article/pii/S0169409X25001334]
- 39. Antiviral strategies against influenza virus - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11825441/]
- 40. 体内功能抗体效应如何定量评估其在抗病毒治疗中的贡献? [https://www.cn-healthcare.com/articlewm/20251119/wap-content-1661053.html]
- 41. and ex In systems at the forefront of vitro modeling and... vivo infection [https://pmc.ncbi.nlm.nih.gov/articles/PMC7172914/]
- 42. Re-evaluation of FDA-approved antibiotics with increased ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10213814/]
- 43. In Vitro vs In Vivo: Complete Comparison + Selection Guide [https://www.assaygenie.com/in-vitro-vs-in-vivo-complete-comparison-selection-guide-research-methods/?srsltid=AfmBOooOSDIxApgjZSXxUvPoBgNMMtGxW5VcRzJfPFbB3AamZdz9xpvx]
- 44. Antimicrobial efficacy validation using in vitro and in vivo ... [https://www.sciencedirect.com/science/article/abs/pii/S0169409X16302587]
- 45. In vitro and in vivo comparison of the anti-staphylococcal ... [https://bmcinfectdis.biomedcentral.com/articles/10.1186/1471-2334-10-153]
- 46. Resource Competition May Lead to Effective Treatment of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3862480/]
- 47. Frontiers | Characterization of the in , ex vitro , and vivo ... in vivo [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2019.00174/full]
- 48. Biofilm antimicrobial susceptibility testing: where are we ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10732061/]
- 49. In Vitro Models of Bacterial Biofilms: Innovative Tools to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10004855/]
- 50. Estimation of Minimum Biofilm Eradication Concentration ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2022.896978/full]
- 51. In vivo verification of in vitro model of antibiotic treatment ... [https://pubmed.ncbi.nlm.nih.gov/7625801/]
- 52. Measuring Antimicrobial Efficacy against Biofilms: a Meta- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6496104/]
- 53. Multi-species biofilms of environmental microbiota isolated ... [https://www.sciencedirect.com/science/article/pii/S2590207524000029]
- 54. Synergistic combinatorial treatments to overcome antibiotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10853682/]
- 55. A Computational Approach for Identifying Synergistic Drug ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005308]
- 56. Improved analysis of in vivo drug combination experiments ... [https://www.nature.com/articles/s41467-025-65218-9]
- 57. Combinational therapeutic strategies to overcome ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1546717/full]
- 58. How to predict effective drug combinations [https://pmc.ncbi.nlm.nih.gov/articles/PMC12152377/]
- 59. Clinical success of anti-infective combination therapy compare ... [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-024-09060-2]
- 60. Do antibiotic combinations proposed from in vitro studies ... [https://revive.gardp.org/do-antibiotic-combinations-proposed-from-in-vitro-studies-lead-to-changes-in-treatments/]
- 61. Study Designs for Evaluation of Combination Treatment [https://pmc.ncbi.nlm.nih.gov/articles/PMC8869609/]
- 62. Systematic review of computational methods for drug ... [https://www.sciencedirect.com/science/article/pii/S2001037022002100]
- 63. In vitro and in vivo analysis of antimicrobial agents alone ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2015.00507/full]
- 64. Humanized Mouse Models for the Study of Infection and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6266013/]
- 65. Ultrasonic irradiation enhanced the efficacy of antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10230259/]
- 66. Humanized Mouse Models for Immuno-Oncology Research [https://www.sciencedirect.com/science/article/pii/S2666364324001516]
- 67. Animal Model Market Size, Share & Growth 2025-2035 [https://www.futuremarketinsights.com/reports/animal-model-market]
- 68. Major updates to FDA-recognized Clinical and Laboratory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12077184/]
- 69. In Vitro vs In Vivo: Complete Comparison + Selection Guide [https://www.assaygenie.com/in-vitro-vs-in-vivo-complete-comparison-selection-guide-research-methods/?srsltid=AfmBOoojxW82WqfHagEYgFTbJcMKOY5UClGSjeaROyw8as1hdloMLumC]
- 70. In vitro vs. In vivo: Is One Better? [https://www.uhnresearch.ca/news/vitro-vs-vivo-one-better]
- 71. Validation framework for in vivo digital measures - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11751004/]
- 72. ICMR's 2025 Guidance on Rapid Diagnostic Validation & ... [https://cliniexperts.com/regulatory-update/guidance-document-on-validation-of-rapid-diagnostics-for-pathogen-identification-and-antimicrobial-susceptibility-testing-ast-2025/]
- 73. Correlation between in vitro and in vivo activity of ... [https://pubmed.ncbi.nlm.nih.gov/1929302/]
- 74. Does In Vitro Potency Predict Clinically Efficacious ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7484912/]
- 75. Impact of Bacterial Biofilm Formation on In Vitro and In Vivo ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC105562/]
- 76. Antibiotics versus biofilm: an emerging battleground in ... [https://aricjournal.biomedcentral.com/articles/10.1186/s13756-019-0533-3]
- 77. Mechanisms of antimicrobial resistance in biofilms [https://www.nature.com/articles/s44259-024-00046-3]
- 78. Correlation between in vivo and in vitro efficacy ... [https://pubmed.ncbi.nlm.nih.gov/2355207/]
- 79. Comparison of the Efficiency of Selected Disinfectants against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10300984/]
- 80. An innovative strategy to treat pathogenic biofilm-associated ... [https://pubs.rsc.org/en/content/articlehtml/2025/md/d5md00776c]
- 81. In Vitro and In Vivo Anti-infective Potential of Thymol ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2021.760768/full]
- 82. In Vitro and In Vivo Assessment of Pharmacokinetic Profile ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11858854/]
- 83. Thieme E-Journals - Drug Research / Abstract [https://www.thieme-connect.com/products/ejournals/abstract/10.1055/a-2061-7074]
- 84. Comparison of Respiratory Microbiomes in Influenza ... [https://www.mdpi.com/1422-0067/26/2/778]
- 85. A novel immune biomarker IFI27 discriminates between ... [https://publications.ersnet.org/content/erj/49/6/1602098]
- 86. Challenges and Opportunities of Nontraditional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5634136/]
- 87. The FICI paradigm: Correcting flaws in antimicrobial in synergy... vitro [https://pubmed.ncbi.nlm.nih.gov/30836058/]
- 88. Menadione as Antibiotic Adjuvant Against P. aeruginosa [https://www.mdpi.com/2079-6382/14/2/163]
- 89. combinatorial use of α-terpineol and colistin [https://pmc.ncbi.nlm.nih.gov/articles/PMC12584673/]
- 90. Methodologies for in and vitro evaluation of in of... vivo efficacy [https://microbialcell.com/researcharticles/methodologies-for-in-vitro-and-in-vivo-evaluation-of-efficacy-of-antifungal-and-antibiofilm-agents-and-surface-coatings-against-fungal-biofilms/]
- 91. Preclinical Studies In Vitro vs In Vivo [https://www.news-medical.net/life-sciences/In-Vitro-vs-In-Vivo-Preclinical-Studies.aspx]